← Back to English Insights
Medical device regulatory insight · 2026-04-13
Global Registration Strategy and Compliance AI Deployment for CGM: Insights from CMEF
CGM Global Registration Strategy and Global Compliance AI Practice

On April 10, 2026, during the Spring CMEF in Shanghai, at the 3rd Continuous Glucose Monitoring (CGM) Technology Seminar,Clinsota founder Feng Huimei delivered a keynote presentation on “Global Registration Strategies, Clinical Compliance Challenges, and AI Solutions for CGM Products.” Drawing on the seminar content and the latest publicly available information, this article seeks to distill the underlying logic and provide practical considerations and recommendations.I. The Global CGM Market: Reshaping the Landscape in a High-Growth Era
1. Market Size and Growth Drivers
The global CGM market was approximately US$14.29 billion in 2025 and is projected to grow to US$35.44 billion by 2032, with a compound annual growth rate (CAGR) of approximately 14.1% from 2026 to 2032. This forecast reflects very strong growth momentum. External market research data also indicate that the global CGM market was approximately US$1.09 billion in 2024 and is expected to grow to US$4.71 billion by 2034, with a CAGR of 16%. Although different reports vary slightly in absolute figures, they all point to the same trend:CGM is a rapidly growing expansion market.Growth is being driven primarily by three factors:- Rising diabetes prevalence: The World Health Organization estimates that the global number of people with diabetes has approached 830 million, the majority of whom are in low- and middle-income countries. This large population provides a long-term demand foundation for CGM.
- Technology iteration and the consumerization trend: The presentation emphasized that CGM is shifting from specialist medical use toward broader consumer health. The emergence of over-the-counter (OTC) products and subscription models has lowered barriers to entry for users. Iterations such as non-invasive/minimally invasive technologies and AI predictive algorithms have improved user experience and accuracy.
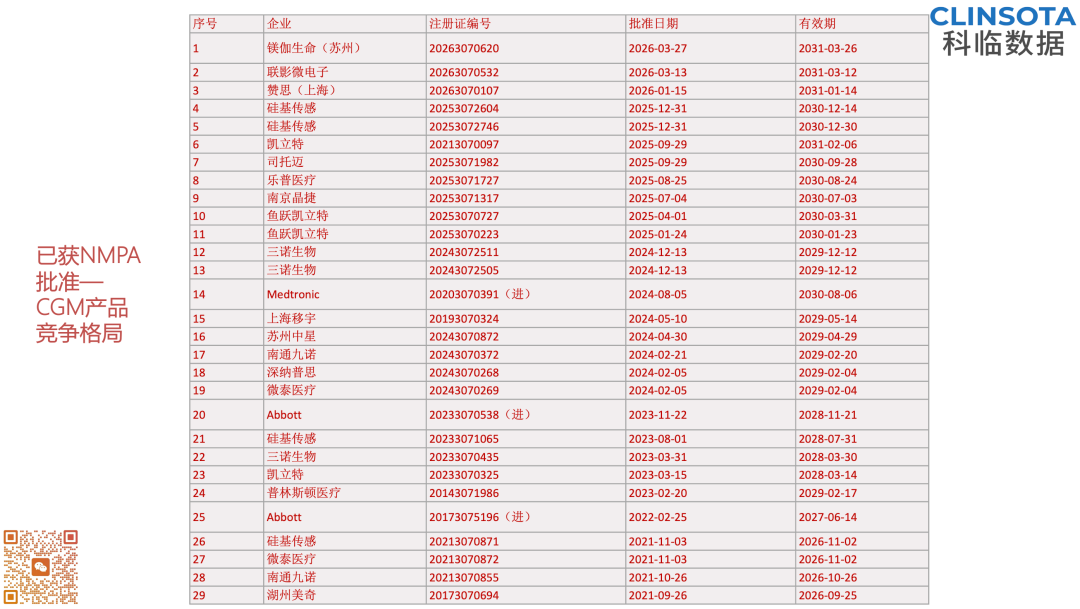
- Reimbursement and policy drivers: An increasing number of health insurance systems worldwide are bringing CGM into reimbursement coverage, driving higher market penetration.Below is an overview of Chinese companies that have obtained NMPA registration certificates to date for reference
2. Competitive Landscape and Market Changes
The market currently shows a clear oligopolistic structure: the three giants—Abbott, Dexcom, and Medtronic—collectively account for nearly 97% of the global market share. However, Chinese domestic manufacturers, represented by Sinocare, Yuwell, SiBionics, and MicroTech Medical, have already increased their global share to 23% and continue to grow through strong price-performance ratios and localized strategies. This indicates that the window of opportunity for later entrants is narrowing,technology and regulatory compliance capabilities will determine whether they can break through.3. Technology Trends and Ecosystem Upgrading
Four major future trends in CGM:- Product consumerization: moving from invasive to minimally invasive and then to non-invasive technologies, such as near-infrared spectroscopy or microneedle technologies, progressively improving user comfort and expanding into broader health populations.
- Scenario diversification: application settings are expanding from the home to hospitals, communities, and remote management, while integrating deeply with insulin pumps and AI algorithms to form closed-loop systems.
- Ecosystem intelligence: AI enablement is upgrading products from data-monitoring tools into intelligent health management platforms that provide predictive alerts and personalized recommendations.
- Business model transformation: subscription models, data-driven services, and ecosystem integration will become the primary revenue models.
These trends indicate that companies must move beyond a hardware-centric mindsetData + algorithms + ecosystemsystem-level thinking to build long-term competitiveness around user experience and health value.II. China-EU Regulatory Differences: Understanding the Fundamental Differences in Logic and Pathways
To achieve global registration, it is essential to fully understand the differences between China's NMPA and the EU MDR in classification, registration unit definition, clinical pathways, and related areas.1. Classification and Registration Units
China: NMPA classifies CGM as a Class III medical device, and its registration unit rules are highly detailed, with emphasis on factors such as sensor design and wear duration. NMPA guidance states that the same registration unit includes the sensor, transmitter, and receiver. If the sensor design or materials differ, or if wear duration differs substantially, they should be divided into different registration units. However, if the sensor design is the same and different wear durations are achieved through software settings, they may be included in the same registration unit. These rules highlight the regulatory impact of hardware differences.EU: In the EU, CGM is mainly classified as Class IIb (and in some cases as Class III, such as closed-loop systems). Classification is relatively flexible and focuses on the device's risk characteristics. The EU does not prescribe registration units with the same level of detail as NMPA, but it emphasizes logical consistency and risk classification. Project experience shows that the EU also accepts claims for different wear durations enabled through algorithm adjustment, such as 7-day and 14-day versions, but they must be clearly distinguished in model designation and intended use.2. Registration Pathway and Clinical Requirements
China: NMPA requires domestic clinical trials. The latest white paper states that all Class II and Class III devices (except those on the exemption list) require clinical data. Clinical data may come from domestic Chinese studies or overseas studies compliant with Chinese GCP, but overseas data must undergo population difference validation, and supplemental bridging studies are required when necessary. NMPA places emphasis on prospective, multicenter, self-controlled study design, requires at least two GCP hospitals, and uses venous plasma glucose as the gold standard.EU: The EU accepts overseas clinical data, but ISO 14155 population GAP analysis is required, and post-market PMCF studies are emphasized. Clinical evaluation relies not only on trial data, but also on literature, experience data, and other evidence, and requires a completebenefit‑risk justification and SOTA (State of the Art) analysis. Under the EU MDR, PMCF is treated as an ongoing process within clinical evaluation, requiring manufacturers to proactively collect real-world post-market data to confirm device safety and performance. In addition, failure to meet specific SOTA and equivalence requirements can lead to submission failure. Many Notified Bodies identify unclear clinical objectives, lack of quantifiable metrics, and insufficient SOTA as the most common issues.3. Key Differences in Clinical Evaluation Focus
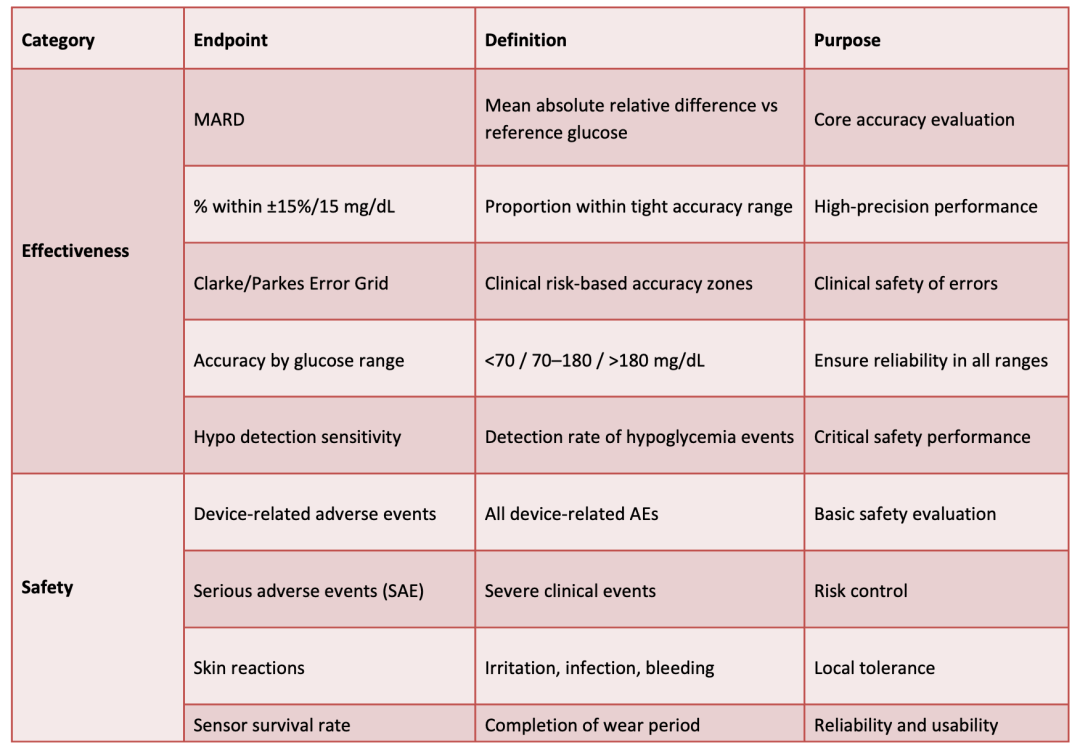
China focuses on data outcomes: NMPA emphasizes prospective, multicenter clinical trial data and specific quantitative metrics, such as 20/20 agreement rate and Mean Absolute Relative Difference (MARD). The key metrics in a China-EU aligned study design include, for example, 20/20% agreement versus venous blood glucose (target ≥60%), Clarke Error Grid Analysis A+B zone proportion (target ≥90%), and MARD ≤20% etc., to demonstrate that this CGM measures accurately, consistently, and safely.The EU focuses on the evidence chain and benefit-risk justification: clinical evaluation must cover SOTA review, equivalence assessment, literature search, clinical investigation, benefit‑risk analysis, PMCF planning, and related elements. Notified Bodies often require clearly defined safety/performance objectives supported by quantifiable thresholds; otherwise, the clinical purpose may be challenged as unclear.III. Dual China-EU Submission Strategy: One Process, One Data Set, GAP Identification for Both Markets
If a company wants to enter both the China and EU markets at the same time, it needs to systematically design a dual-submission strategy of “one dossier set, two-country submission.” Three core strategies are summarized:Registration Unit Alignment
: define shared China-EU requirements during the product design stage. Use software algorithms to control different wear durations so that the same hardware platform can meet different market needs, while clearly distinguishing model designation and intended use. This requires the R&D, regulatory, and clinical teams to participate in compliance planning from the design source.Global Multicenter Clinical Trial
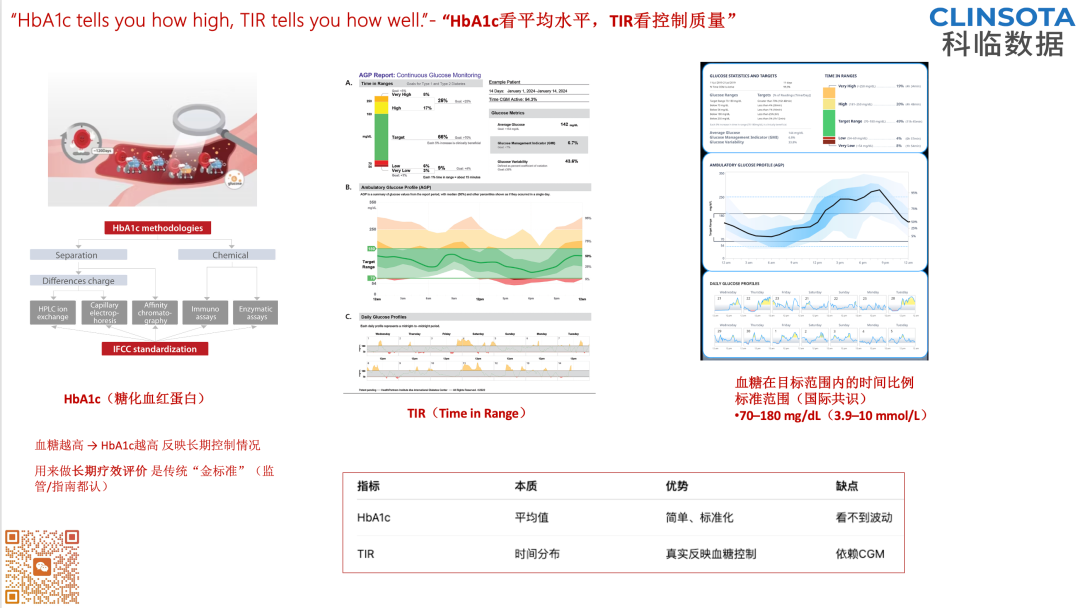
: design a prospective, multicenter, self-controlled global clinical trial so that one data set can simultaneously satisfy both China and EU requirements. Sample size must take into account the statistical requirements of both jurisdictions. Primary endpoint metrics should simultaneously meet 20/20 agreement rate, MARD, and related requirements; the protocol, inclusion/exclusion criteria, and evaluation metrics should align with the EU's SOTA and GSPR approach.Common clinical endpoints include, but are not limited to, the following:For overseas clinical trials, an ISO 14155 GAP analysis and a PMCF plan are also required, taking into account population differences such as skin color, BMI, genetics, etc., as well as HbA1c(glycated hemoglobin) and TIR(Time in Range) considerations.Shared systems and documentation
: QMS documentation (ISO 13485), risk management reports (ISO 14971), software validation documentation (IEC 62304), and similar system documents can be shared; test reports such as electrical safety (IEC 60601‑1), electromagnetic compatibility (IEC 60601‑1‑2), and biocompatibility (ISO 10993) may also be mutually recognized between China and the EU. At the same time, device-specific materials such as Instructions for Use (IFU), labeling, and clinical evaluation documentation (GSPR/CER) that comply with local language and regulatory requirements must be prepared. Through careful management of shared materials and differentiated materials, registration efficiency can be substantially improved.In practice, a dual-filing strategy requiresparallel project initiation, alignment of key milestones, and timeline estimation; parallel execution can significantly save time. Companies should develop a detailed project plan that defines the deliverables and timelines for stages such as design freeze, clinical study initiation, regulatory submission, and post-market surveillance.IV. Clinical Compliance Challenges: The Complexity of Evidence Engineering
Clinical compliance is not just about drafting reports; it is a highly complexevidence engineering. The main challenges include:- Literature searching and State of the Art (SOTA) development are cumbersome: the EU emphasizes the need to establish a comprehensive SOTA covering the current therapeutic landscape, alternatives, endpoints, and more. Manual literature searching, screening, and classification are extremely time-consuming and can easily miss key evidence.
- Equivalence assessment is challenging: EU MDR requires demonstration of equivalence between the subject device and the comparator device across technical characteristics, biological characteristics of materials, and clinical characteristics. Insufficient justification may lead to a requirement for additional clinical investigations.
- Cross-document consistency management is complex: documents such as the Clinical Evaluation Report (CER), Clinical Evaluation Plan (CEP), PMCF Plan, and GSPR matrix are interrelated. When managed manually, any change can create inconsistencies across other documents and increase the risk of rework.
- The regulatory environment changes rapidly: risk management, PMCF, and Periodic Safety Update Report (PSUR) obligations under EU MDR are ongoing; China also continuously updates technical requirements and guidance. Companies must keep their documentation up to date and maintain compliance.
- Talent and cost pressures: high-quality compliance teams are scarce, and compliance cycles are long and costly, posing a particular challenge for start-ups.
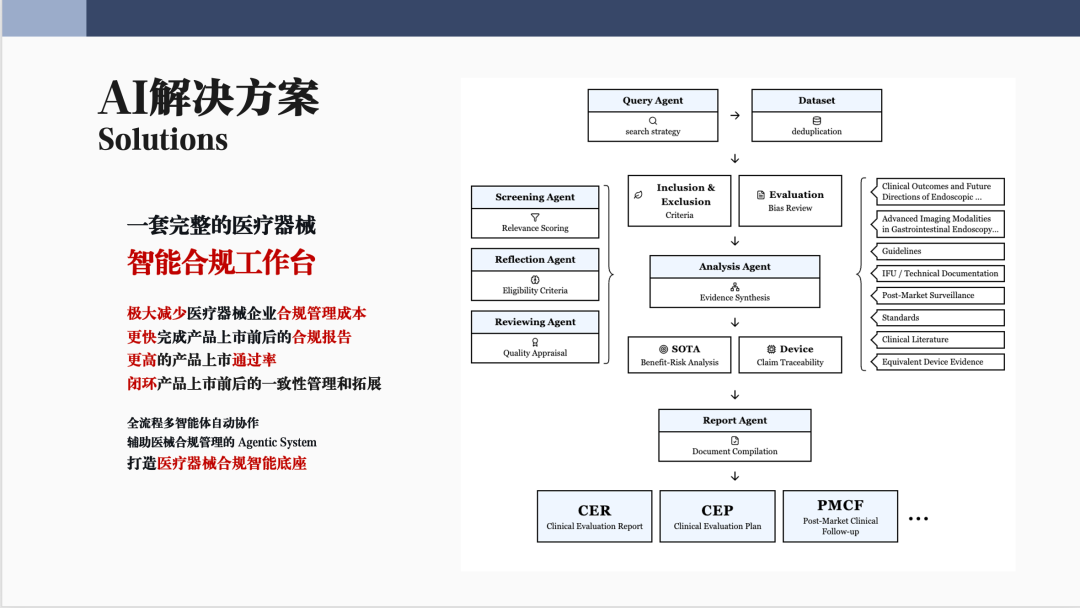
V. AI Compliance Platform: Practical Implementation of Multi-agent Systems
To address complex compliance challenges, Clinsota has developed a "multi-agent (Agentic) medical device compliance platform." Unlike general-purpose generative AI, this platform is focused on medical device compliance use cases and has the following features:1. Multi-agent Collaboration
The platform uses multiple specialized agents working in coordination, including literature retrieval agents, screening agents, analysis agents, and report-generation agents. Through task decomposition and automated collaboration, it enables end-to-end automation from retrieval, screening, and extraction to analysis and report generation.2. Closed-loop Evidence Chain and Full-output Alignment
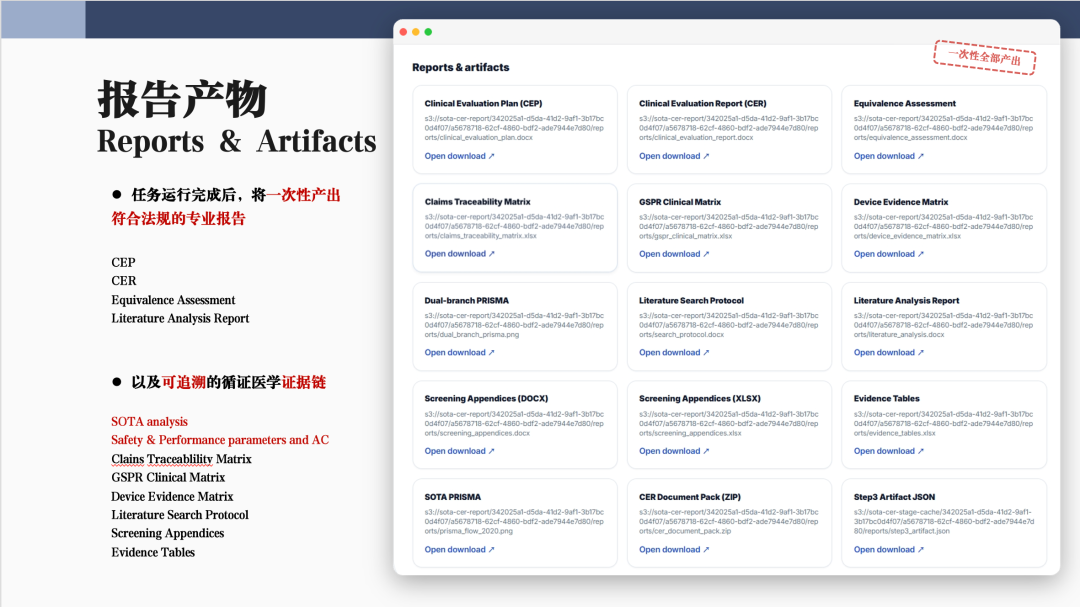
Upon completion of a task, the platform generates in a single run professional reports that comply with MDR and NMPA regulatory requirements, such as CEP, CER, Equivalence Assessment, and literature analysis reports, while also producing a traceable evidence-based medicine evidence chain. In the form of an evidence chain matrix, it maps SOTA analysis, benefit-risk, clinical claims, and literature sources one by one, establishing Claims Traceability Matrix, GSPR Clinical Matrix, etc., so that all content is fully traceable.In addition, the platform supports full-output alignment, meaning that any revision to one report can be automatically synchronized across other related documents to ensure overall consistency. This “four-dimensional alignment” covers clinical compliance reports, jurisdiction-specific regulatory requirements, evidence-based literature, and user device documentation, thereby creating a unified compliance knowledge base.3. Automation of Equivalence Assessment
Equivalence Assessment is a key feature of the platform. In accordance with MDR requirements, the system compares the subject device with comparator devices across three dimensions: technical characteristics (design principles), biological characteristics (materials and substances in contact with the human body), and clinical characteristics (intended clinical use and user population). In addition to analyzing similar-device materials uploaded by the user, the system automatically retrieves external literature for substantiation and performs joint evaluation through multiple reasoning agents. This can significantly improve first-pass regulatory submission success rates and help determine whether new clinical investigations may be waived.4. Cost Reduction, Acceleration, and Implementation Recommendations
Using an AI compliance platform can deliver practical value in the following areas:- Time savings: Automated retrieval and report generation significantly reduce manual effort and accelerate completion of compliance reports.
- Lower costs: Reduce the size of the compliance team, shorten the registration timeline, and avoid rework caused by document inconsistencies.
- Higher quality: A complete evidence chain and automated alignment mechanisms reduce the risk of deficiency letter responses from the Notified Body or NMPA.
- Empowering SMEs: Startups often have limited staff; with the platform, they can rapidly build compliance capabilities and stay focused on technological innovation.
VI. Future Trends and Implementation Recommendations
1. Technology Evolution and Market Expansion
Non-invasive measurement, extended wear duration, automatic calibration, and closed-loop insulin delivery will be the main directions for future products. AI will not only be used to predict glucose trends, but will also integrate personalized treatment plans and expand into the field of digital therapeutics. As smartphones and wearable devices become more widespread, CGM will gradually become part of broader health management platforms.In the market, North America still holds the largest share, but the Asia-Pacific region, especially China, is growing the fastest. Domestic companies have localized advantages in algorithms and application ecosystems, and can differentiate themselves through cost-effectiveness, precise prediction, user experience, and service ecosystems.2. Regulatory Trends and International Collaboration
The EU MDR will continue to update regulatory guidance after 2025, strengthening PMCF and risk management requirements. In China, the NMPA has further refined registration unit delineation, equivalence assessment, and clinical requirements through documents such as the Guiding Principles for Continuous Glucose Monitoring Systems (2023). These regulations show that regulators encourage innovation, but emphasizesafety and scientific evidence. Companies should closely monitor regulatory updates and leverage international collaborative data and real-world evidence to support regulatory submission.3. Recommendations: Compliance by Design, Data as Competitive Advantage
1.Embed compliance thinking at the design stage: From the outset of product development, companies should consider the differences between Chinese and European regulations and design flexible registration units, intended use statements, and algorithm strategies.2.Build a global clinical strategy: Plan multicenter studies early, appropriately control sample size and endpoint selection, and ensure the data can support submissions in multiple jurisdictions. Pay attention to differences across populations, BMI, skin tone, and other factors.3.Digitalized compliance management: Adopt AI-powered compliance tools and data management platforms to enable evidence-chain management and alignment across all deliverables, reducing the risk of rework.4.Cross-functional teams and continuous learning: Regulatory, R&D, clinical, and AI teams should work collaboratively, regularly review regulatory updates, Notified Body feedback, and clinical data performance, and adjust strategy in a timely manner.5.Focus on business model innovation: Beyond hardware sales, explore subscription services, data analytics, and remote health management to generate recurring revenue.Conclusion
As CGM evolves from a medical device into a health management platform, the global regulatory environment is becoming increasingly stringent and complex. Clinsota’s perspective treats registration compliance as an engineering problem and emphasizes buildingreusable, traceable, and scalablesystems across product design, clinical strategy, and compliance workflows. The analysis in this article also suggests to companies:·The market is not a red ocean, but a period of rapid growth and structural reshaping, giving domestic companies an opportunity to overtake through algorithmic and ecosystem capabilities.·Chinese and EU regulatory frameworks reflect different underlying logic: China emphasizes data outcomes, while the EU emphasizes the evidence chain and risk justification. Understanding these differences and designing a unified strategy that satisfies both sides is the key to successful dual-market submission.·Compliance digitalization is the future trend: Multi-agent AI platforms can substantially improve efficiency in literature searches, equivalence assessment, and report generation, but they must be used under the guidance of a professional team.Global registration is no longer just about obtaining approval; it has become part of corporate strategy. Only by understanding market trends, carefully designing products, digitalizing compliance workflows, and establishing cross-functional collaboration mechanisms can companies stand out in the next round of competition.The above is part of Clinsota's sharing on global registration strategy for CGM and practical applications of AI in global medical device compliance. We welcome further discussion and exchange with industry peers.