

1. Background to the delegated act
The background to the delegated act is that well-established technologies (WET) are relatively simple devices with common and stable designs, limited variation, and well-knownsafety and clinical performance characteristics, as well as a long market history . Criteria for identifying WET have been established in the Medical Device Coordination Group guidance (MDCG 2020-6). Article 61(6)(b) of Regulation (EU) 2017/745 lists a series of technologies,which, as WET, may benefit from a simplified approach under which the requirement to conduct a clinical investigation may be waived . In addition,Article 61(8) empowers the Commission to extend the WET list to devices similar to those listed . Based on the experience accumulated with the MDR to date,this delegated act aims to expand the list of device types set out in Article 61(6)(b) of Regulation (EU) 2017/745 so that they are exempt from the requirement to conduct clinical investigations.
2. Procedure prior to adoption of the act
The list of WET devices included in this delegated act was developed in 2025 through extensive consultation with relevant stakeholders, including observers from the MDCG Clinical Investigation and Evaluation / Performance Study and Evaluation Working Group and the Borderline and Classification Working Group, as well as competent authority members of the MDCG itself.
3. Legal elements of the delegated act
This legal act is intended to expand, under Article 61(6)(b) of Regulation (EU) 2017/745,the list of types of implantable devices and Class III devices ,that are exempt from the requirement to conduct clinical investigations.

4. Core considerations for determining WET status:
A common, simple, and stable design
Well-established safety and verified clinical performance
Well-known clinical performance characteristics, as reflected in the evolution of standard-of-care indications and the State of the Art;
A long history of use in the Union market
Consistency with the State of the Art
5. Expansion of the scope of WET devices under this regulation
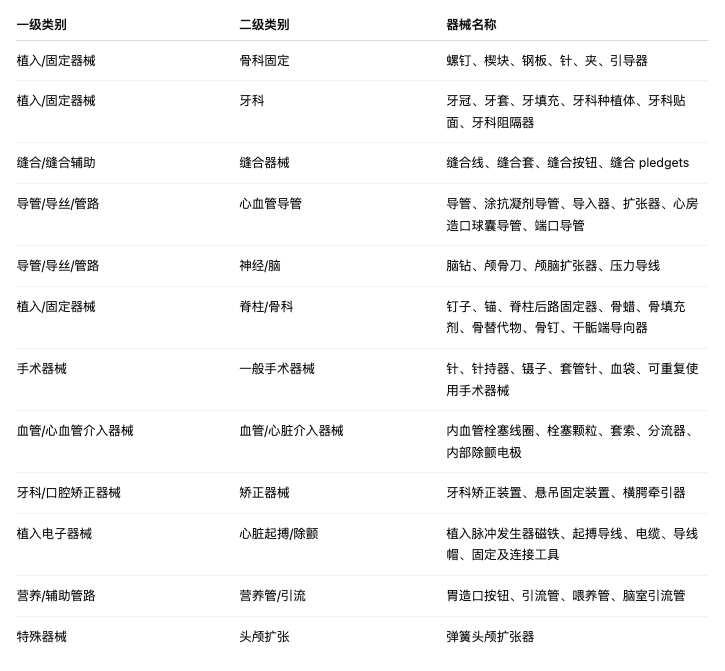
This Regulation shall enter into force on the twentieth day following its publication in the Official Journal of the European Union. This Regulation shall be binding in its entirety and directly applicable in all Member States.
6. Analysis of regulatory trends
7. Considerations for corporate clinical compliance strategy
Evidence-based medicine, PMCF data, registries, RWE (real-world evidence), and clinical evaluation (CER)will be further elevated, and dataanalysis will need to be stronger, more systematic, and more standardized, shifting the focus from “whether clinical investigation data exist” to “whether the data are credible, complete, and traceable.”
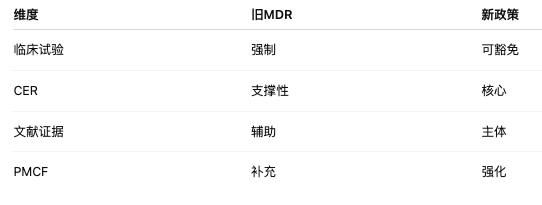
8. Conclusion or impact
The EU regulatory framework is expected to shift comprehensively from a “clinical investigation-driven” model to an “evidence-driven” model. A company’s core capability will no longer be simply “conducting trials,” but rather building a high-quality clinical evidence system.