← Back to English Insights
Medical device regulatory insight · 2026-05-19
EU 2026/977: Analysis of the New Notified Body Rules—Effective from 25 February 2027, Major Changes to Medical Device CE Certification Requirements
This article introduces the new rules emerging in the EU medical-device compliance market for more standardized and transparent collaboration among multiple parties — rule-makers, regulators, regulated entities, and implementers:
Since the publication of the MDR and IVDR in 2017, NB resource sho

This article explains, within the EU medical device compliance market, the new rules for rule-making parties, regulators, regulated parties, and implementing parties as all stakeholders move toward more standardized, transparent, and collaborative practices:Since the MDR and IVDR were issued in 2017, three issues have hung over every manufacturer: insufficient Notified Body resources, inconsistent review standards, and unpredictable certification timelines. MedTech Europe has repeatedly called for greater predictability and transparency, and Hogan Lovells has also pointed to “markedly different certification practices” among Notified Bodies, with SMEs under the greatest pressure.In May 2026, the European Commission formally issued Implementing Regulation (EU) 2026/977, establishing unified rules for Notified Bodies’ conformity assessment activities for the first time. This is not a minor adjustment, but an end-to-end framework governing quotations, timelines, and interruption mechanisms.Clinsota has distilled the key changes in this regulation into the quick-read cards below—each one addresses a question every medical device manufacturer must clearly understand.I. The three systemic pain points facing Notified Bodies after MDR/IVDR implementation
This regulation did not emerge in a vacuum.In the years since MDR/IVDR took effect, three problems have become increasingly serious. First, Notified Body resources have been severely constrained, stretching certification schedules to unacceptable lengths, with the IVDR backlog especially striking. Second, review standards vary widely across Notified Bodies: the same technical documentation can trigger completely different deficiency requests depending on the organization, leaving manufacturers unsure how to respond. Third, certification timelines have been highly unpredictable, and the “clock-stop” mechanism has been used so frequently that companies cannot build reliable market access timelines.Smart MDR noted in its analysis: “This regulation is intended to addresslong-standing concerns such as inconsistent Notified Body practices, unpredictable timelines, differing quotation methodologies, and divergent approaches to recertification.” That sentence captures the legislative intent of the regulation as a whole—it is not a marginal enhancement, but an inevitable direction of travel.Source: MedTech Europe / Hogan Lovells analysisII. Two key dates—do not confuse them
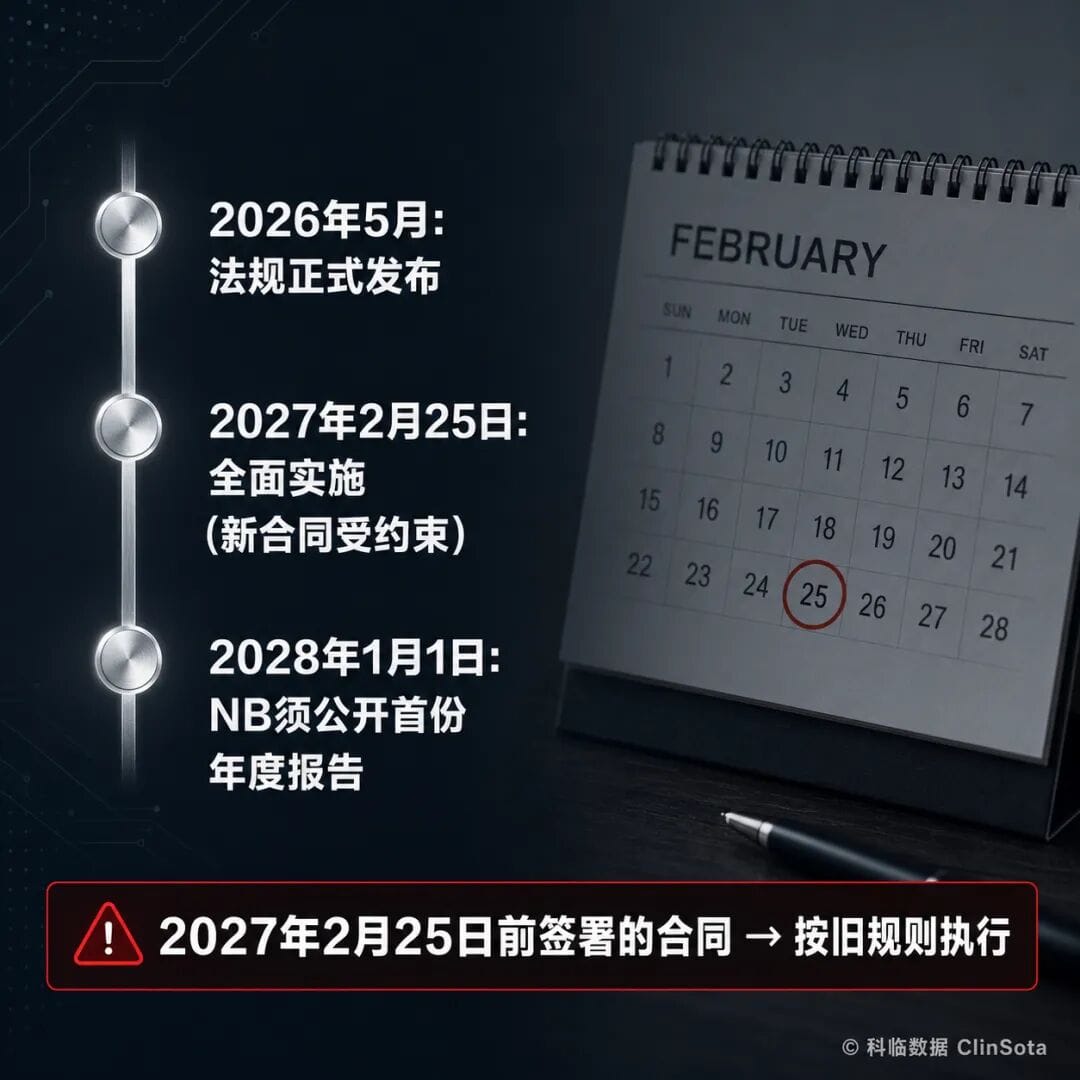
The transition period has two critical dates, and confusing them can be costly.25 February 2027 is the full implementation date. From that day onward, all new conformity assessment contracts signed between Notified Bodies and manufacturers will be governed by the new rules. Contracts signed before that date will continue under the previous rules.1 January 2028 is the other key date. From that day onward, every Notified Body must publish its first annual business report, including certification volumes, timelines, and fee ranges.What does this mean? If you are preparing a CE certification application, signing the contract before 25 February 2027 versus after that date places you under two entirely different regulatory frameworks. The timing of that window directly affects your cost and timeline.In one sentence: contracts signed before 25 February 2027 → follow the previous rules.III. Full quotation transparency—an end to “shock invoices” and hidden charges
Under the old model, Notified Body quotations were often opaque. Fee structures lacked transparency, price increases were arbitrary, and companies often learned the true cost only when the invoice arrived.The new model puts all of this on the table. As Hogan Lovells observed: “For manufacturers, this means significantly greater cost predictability and transparency from the very start of the conformity assessment process.”How exactly is this made transparent? Three mandatory requirements:First, before issuing a quotation, the Notified Body must collect 10 categories of company information: basic company details, SME status, authorized representative information, manufacturing sites, key suppliers, device description, intended purpose, risk classification, conformity assessment procedure, and the change plan and recertification scope.Second, the quotation must break down fees into two main components—QMS assessment and technical documentation assessment—and must also include typical fees for surveillance audits and unannounced audits.Third, any fee increase above 10% must be notified in advance and justified.For companies, this means that for the first time you can see a clear basis for comparison before selecting a Notified Body.IV. The first-ever “hard timeline”—certification can no longer be delayed indefinitely
This is the most consequential change for manufacturers in the entire regulation. For the first time, the Regulation sets a maximum timeframe for each stage of conformity assessment:- Application review and contract signature: 30 days (from receipt of a complete application)
- QMS audit: 120 days (from the first activity under the audit program)
- Technical documentation assessment (product verification): 90 days (from the start of the technical documentation assessment)
- Decision and certificate issuance: 20 days (from the day after completion of the final QMS or product verification)
The clock starts on the date the Notified Body begins the first activity of the relevant stage, not on the date the application is submitted. This detail matters—if you have submitted the materials but the NB has not yet started the review, the clock does not start.Major change assessments are also subject to deadlines: 30 days to review the change proposal, 90 days for any additional conformity assessment, and 20 days to issue the certificate supplement.Smart MDR makes one important point: “The new framework may improve predictability, but it does not guarantee a faster overall certification timeline in every case.” This is because the Regulation allows timelines to be interrupted in a range of circumstances, and interruption periods do not count toward the clock.V. A cap on interruptions—“clock-stop” can no longer be extended indefinitely
The interruption mechanism (clock-stop) has been the biggest driver of prolonged certification delays. Mid-review, the NB would identify missing materials, pause the clock, request supplementary information, and then resume once it was submitted—this cycle could repeat over and over, leaving manufacturers with no leverage.The new rules set strict caps for each stage:- Application review: maximum 1 interruption
- QMS audit: maximum 4 interruptions (plus 2 additional interruptions for each extra site in a multi-site setup)
- Product verification: maximum 4 interruptions
- Decision and certificate issuance: maximum 1 interruption
This means piecemeal submissions will consume your interruption allowance. The old strategy of “submit a first version, wait for the NB to find issues, and then supplement gradually” will no longer work. The quality of the initial submission now determines certification efficiency—this is not a slogan, but the operational logic of the new rules.There is also a safeguard for manufacturers: exceeding the maximum timeframe or using the interruption mechanism cannot, by itself, be the sole ground for refusing certification. The assessment must be completed on the merits. If an NB tries to reject the file simply because “time is up,” you have grounds to challenge it.VI. Where NB certification fees are headed—higher in the short term, more rational in the medium to long term
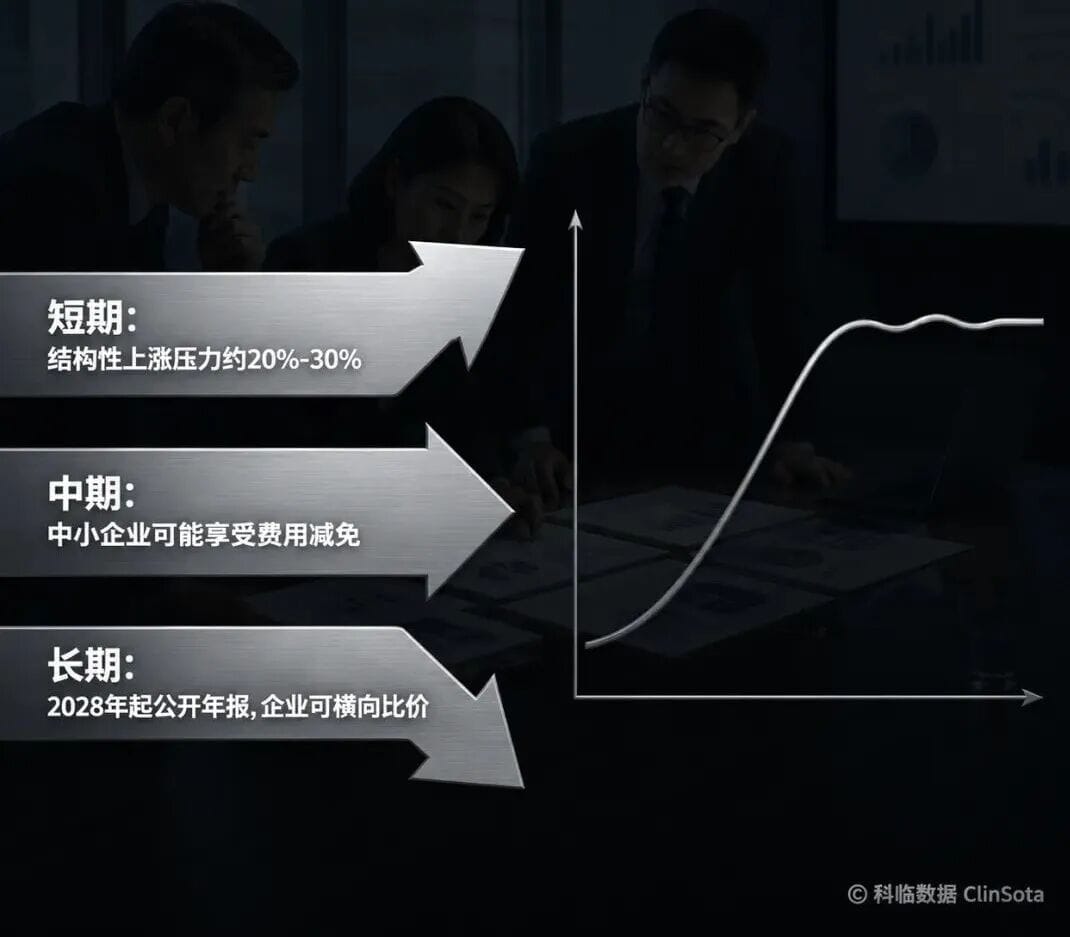
Fees are everyone’s top concern, and the conclusion should be viewed from three angles.In the short term, structural upward pressure is real, at roughly 20%–30%. The reason is that the Regulation imposes higher quality management and transparency requirements on Notified Bodies, increasing their own compliance costs; rigid constraints on personnel, time, and accountability are pushing costs upward across the full chain.In the medium term, SMEs may benefit from mandatory fee reductions. Osborne Clarke notes that the MDR/IVDR amendment draft would require NBs to apply fee reductions for SMEs in order to make certification more affordable. However, Team-NB has strongly objected—54% of small and micro device manufacturers in the EU market come from outside the EU, so the debate continues over whether mandatory reductions would truly protect EU-based companies.In the long term, fees are likely to become more rational. From 2028 onward, each NB must publish an annual report including certification volumes, timelines, and fee ranges. This means manufacturers will be able, for the first time, to make meaningful comparisons across NBs; transparency itself will drive competition and restrain unreasonable price premiums.VII. Different certification stages require different response strategies
Whatever the regulatory changes, the ultimate question you need to answer is: what should I do with my product now?Scenario A: certification already in progress: If you have already signed a contract with an NB but certification is not yet complete, the current contract remains governed by the previous rules. However, you should proactively communicate with the NB and, where possible, align progress with the new framework—at a minimum, you should set expectations internally against the new timelines.Scenario B: planning to apply soon: This is the most affected group. Three recommendations: first, move quickly to use the window before 25 February 2027—if you can sign the contract before that date, you will not be subject to the new timeline and quotation rules; second, complete a technical documentation completeness check before submission, because interruptions are limited under the new rules; third, obtain quotations from multiple parties and use the standardized quotation structure to compare them side by side.Scenario C: holding certificates under the old Directives: These may continue to be used during the transition period, but only if the strict conditions of MDR Article 120 are met, and no significant changes are made to the product. One legislative development worth watching is the possibility that CE certificates may no longer have a fixed 5-year validity period and could become permanently valid—this is still under discussion and should be monitored.VIII. Four urgent actions for medical device manufacturers
To distill all of the above analysis, these are the four things you should do now.First, review your contract signature timing. Determine the expected application timing for each product and assess whether contract signature can be completed before 25 February 2027. If you can meet the window, accelerate progress; if not, prepare early in line with the new rules.Second, conduct a technical documentation completeness review. Under the new rules, interruptions are capped and opportunities to submit supplementary materials are limited. Check each item against MDR/IVDR Annex II and Annex III, ensure the Clinical Evaluation Report (CER) is prepared in accordance with the latest MDCG guidance, and confirm that the QMS documentation covers all Article 10 requirements. An internal or third-party pre-assessment before formal submission is recommended to reduce the number of deficiency cycles during NB review.Third, compare the public annual report data of NBs. From 1 January 2028 onward, proactively review each NB’s annual report, focus on median certification timelines and fee ranges, and incorporate transparency performance into your NB supplier evaluation.Fourth, urgently focus on 26 May 2026. This is the deadline for the MDR transition period for Class III custom-made implantable devices. After this date, they can no longer be marketed in the EU on the basis of MDD certificates. If your portfolio includes such products, this is not something merely “worth watching”—it is a mandatory action.Professional compliance support: Clinsota | End-to-end medical device CE certification servicesIn closing
Implementing Regulation (EU) 2026/977 is not the end point. MedTech Europe has made this clear: the Regulation is a “complementary stabilizing measure” that needs to advance in parallel with broader MDR/IVDR revisions, and “both are necessary; neither is sufficient on its own.”For manufacturers, this Regulation brings a more predictable and transparent certification environment. But predictability does not automatically mean faster timelines, and transparency does not automatically mean lower costs. The rules have changed, and your preparation cadence must change with them.As MedTech Europe CEO Oliver Bisazza said:“This is a meaningful first step toward a more predictable and more consistent system; what matters now is effective implementation.”