What exactly is Route B?

Adjusting inclusion criteria Updating the informed consent form version Adding two investigators
Average approval time:7 days Automatic approval rate:89% Stopped rate:11%(mainly due to incomplete materials)
Which projects can use it? Which cannot?
Changes to visit frequency (for example, from 3 visits to 5 visits) Minor adjustments to the recruitment strategy (e.g., broadening the age range) Changes in Ethics Committee membership Wording refinements to the informed consent form Updates to the data management plan
Changes to the primary endpoint Changes to device design or specifications Adjustments to procedures related to subject safety Major revisions to core parts of the protocol
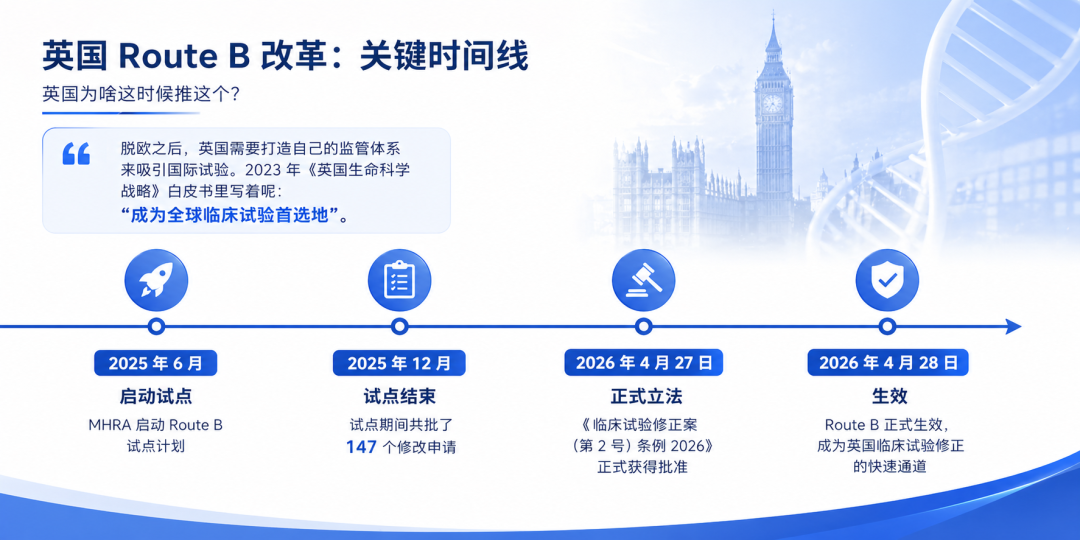
Why is the UK introducing this now?
June 2025: Pilot launched December 2025: Pilot concluded, with 147 amendment applications approved 27 April 2026: Formally enacted 28 April 2026: Took effect
U.S. FDA: there is a Breakthrough Devices pathway, but the overall timeline remains long, averaging 12-18 months European Union: CTIS provides a unified 60-day assessment, but the process is complex and often exceeds the timeline Japan PMDA: Its early consultation mechanism is strong and suitable for innovative devices, but the documentation requirements are detailed
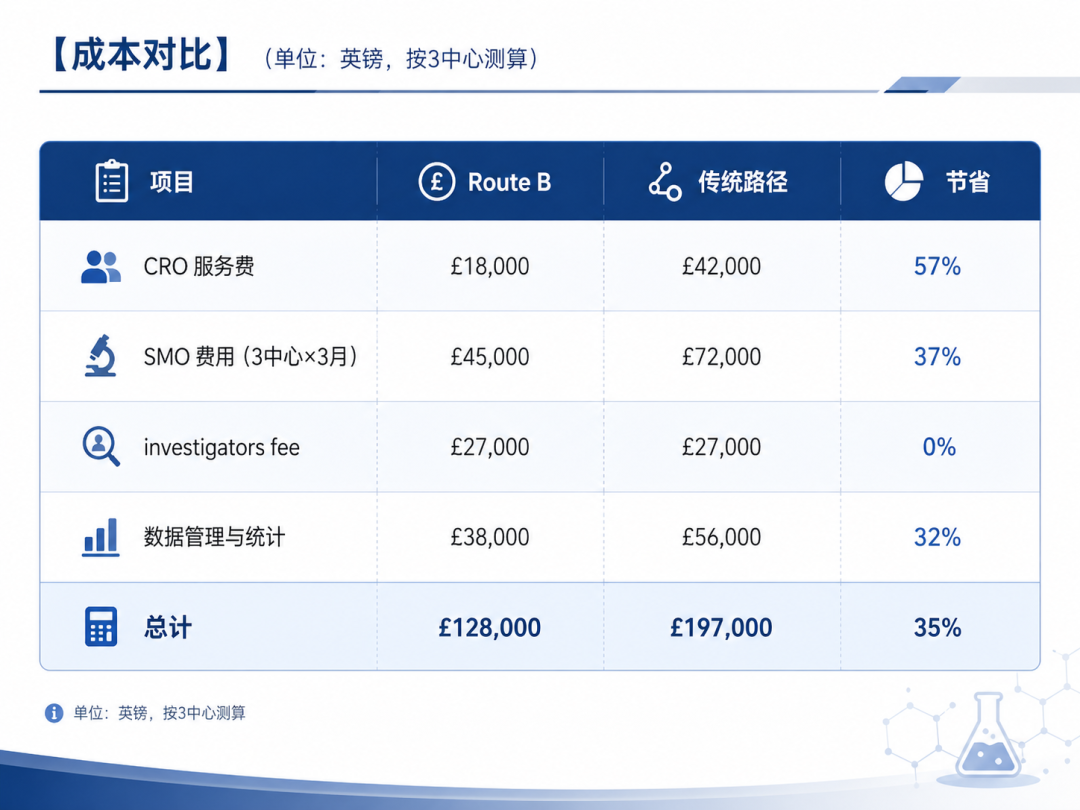
What is the true cost of running a UK project?
**Upfront pre-submission review**: Have your internal team or a consultant review the dossier first to avoid an MHRA hold. We recommend budgeting £5,000-£10,000 for the pre-review. **Communication cost**: MHRA may raise questions, and you will need to respond within 24-48 hours. If your team is outside the UK, time-zone differences with the London office can be an issue, so it is best to have a local contact.
How can smaller companies control these costs?
The core process (submission and follow-up) can be outsourced, but ethics committee communication should be handled in-house as much as possible Use our template checklist for self-assessment to save on consulting fees Pay in stages: 30% upon proposal approval, 40% upon MHRA acceptance, and 30% after approval
What should you do if MHRA places the study on hold?
In the pilot, 11% were placed on hold, mainly for the following reasons:
Incomplete ethics documentation (40%) Issues with the informed consent form (30%) The SAE reporting process was not clearly described (20%) Protocol deviations were not documented (10%)
Hold an internal meeting within 24 hours to assign who will address each gap Submit the additional materials within 72 hours (if you delay too long, the MHRA may view it as a lack of engagement). If the MHRA has not responded within 15 days after the resubmission, proactively call to follow up. A 15-day grace period is generally granted.
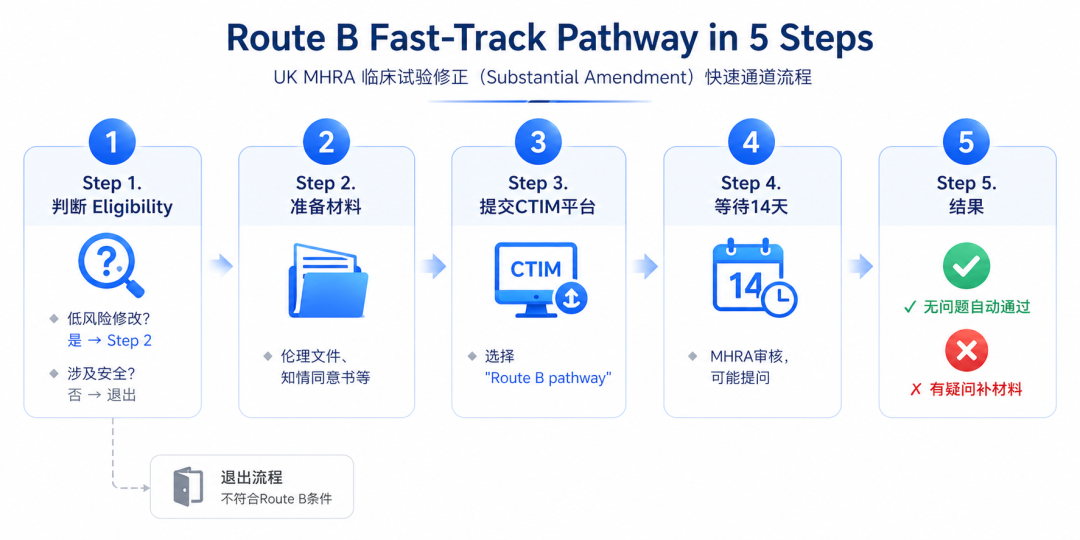
For your own project, can it actually proceed via Route B?
I have compiled 5 questions for you to review first:
**Does the change affect subject safety?** → No **Does it qualify as a low-risk modification?** (recruitment, informed consent, personnel) → Yes **Can all materials be submitted in full at one time?** → Yes **Has Ethics Committee approval already been obtained?** → Yes **Can you respond to MHRA questions within 14 days?** → Yes
Class III devices require particular caution.
Amendments involving device design changes are generally not eligible for Route B. However, changes such as adjusting visit frequency or relaxing enrollment criteria may be eligible.
About 70% of amendments are eligible for Route B. IVD reagents are particularly well suited.
Minor version iterations (for example, fine-tuning a detection algorithm) may proceed via Route B. Major version updates or retraining a machine learning model may require re-approval.
For joint submissions, the MHRA must coordinate with the EMA. It is advisable to begin discussions 6 months in advance
Let’s get practical: what do you need to prepare?
Ethics Committee approval documentation (version and date must match) Informed Consent Form (latest signed copy) SAE reporting procedure (clearly specify the 24-hour contact person) Protocol deviation log (for the most recent 3 months) Data Management Plan (including the data lock timeline) Investigator qualification documents (GCP certificates, CVs) Insurance certificate (clinical trial liability insurance) Medical device clinical trial filing documentation (if applicable)
What stage is your project at?
Started, currently looking for a CRO In planning, evaluating the protocol Still observing, waiting a bit longer Not considering it, focused on the domestic market
A few final remarks
#MedicalDevices#ClinicalTrials#CRO#GlobalMarketAccess#PolicyAnalysis
