

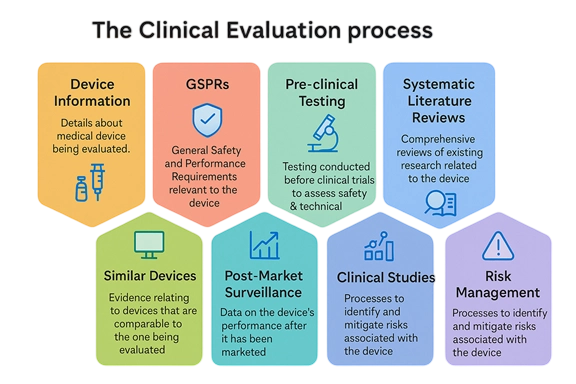
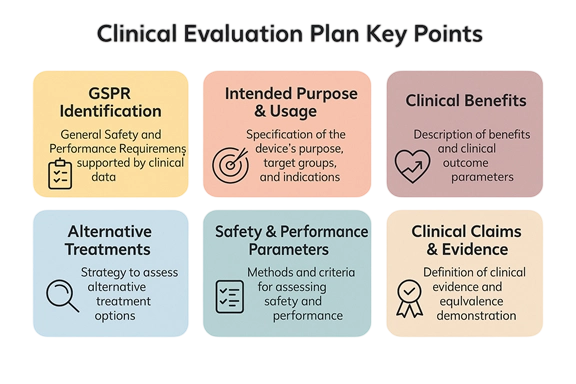

临床评价与 CER / CEP
围绕 MEDDEV / MDR Article 61 / MDCG 要求,组织文献检索、等同性与临床论证。
EU
FDA
NMPA
CER
QA
PMCF
EvidenceGap → Route
CLINICAL LIFECYCLE MANAGEMENT
先判断当前属于哪一种关键任务
把服务入口改成医疗器械团队正在面对的真实决策点:临床评价、审评回复,还是多市场注册复用。
Clinical
临床证据能不能支撑申报
判断临床评价缺口
Response
发补和审评问题怎么回复
建立回复框架
Market
多市场注册路径如何复用
复用资料与证据资产
CORE CRO DOMAINS
围绕同一份证据资产,组织六类核心工作。
不把服务拆成孤立文件,而是按医疗器械企业真正推进注册、审评与上市后维护的顺序组织。
CE MDR / IVDR 技术文档路径
把 GSPR、CER、风险管理、PMS / PMCF 与公告机构关注点串成同一条证据链。
FDA 510(k) 与 AI Response
判断 predicate、substantial equivalence、性能测试和临床数据触发条件,并准备 Additional Information 回复材料。
NMPA 注册与同品种临床评价
处理注册路径判断、同品种证据组织、资料补正与审评意见沟通。
PMCF / PMS / PSUR 上市后闭环
让上市后证据持续反哺 CER 更新、残余风险判断和多市场维护策略。
发补 / 不符合项 / Deficiency Letter
把 NMPA 发补、FDA AI、NB Deficiency Letter 与体系不符合项转成可关闭的响应路径。
DEFICIENCY LETTER RESPONSE
把发补和不符合项,转成可关闭的证据路径。
面对 TÜV / NB Deficiency Letter、FDA Additional Information、NMPA 发补或体系不符合项,先区分“资料缺失、证据不足、逻辑不闭环、测试/临床触发”四类问题,再组织回复。
RESPONSE FRAMEWORK72H / TRIAGE
逐条拆解审评问题识别法规条款、审评关注点、证据缺口和不可直接承诺的风险。
重组临床与技术证据把 CER/CEP、等同性、测试、PMS/PMCF 与风险管理重新串成论证链。
输出可递交回复包形成回复矩阵、补充资料清单、责任分工与下一轮沟通口径。
NB / TÜV Deficiency Letter
FDA Additional Information
NMPA 发补
体系不符合项
WHAT YOU RECEIVE
一次沟通后,您应该得到什么
不是一份泛泛报价,而是一组可以让团队继续推进的判断。
CLINSOTA / OUTPUT01—03
01
证据缺口判断当前资料哪里足够,哪里会被审评追问。
02
下一步路径建议先补临床证据、先回复发补,还是先确定市场路径。
03
资料行动清单下一轮应准备的文件、证据和回复材料。
医疗器械 CRO 常见问题
医疗器械 CRO 可以为企业提供哪些支持?
临床评价、CER/CEP、全球注册资料、PMCF/PMS/PSUR 与审评问题响应。
CER 和 CEP 一般需要多久完成?
常规项目通常为 4–12 周,取决于产品风险等级、已有证据和目标市场要求。
欧盟 MDR 临床评价一定需要临床试验吗?
不一定,需要结合器械类型、风险等级、等同性证据和公告机构关注点判断。
FDA 510(k) 是否需要临床数据?
多数 510(k) 以实质等同比对和性能测试为核心,但部分产品需要临床数据或临床论证。
MDR Article 61 相关问题需要先做什么?
先判断是否能通过等同性、文献、已有临床证据和 PMCF 计划建立临床评价闭环,再决定是否补临床研究。
NMPA 注册资料被补正时,先补哪一类内容?
先拆解审评意见对应的证据缺口,再区分同品种评价、临床评价、技术测试、风险管理或体系文件补充。
SEND THE CURRENT NODE
把当前节点发给我们。
产品类型、目标市场、资料状态或审评问题,都可以作为开始。
产品类型
目标市场
当前问题
