


EU 2026/977:NB新规解读——2027年2月25日起实施,医疗器械CE认证规则迎来重大变化

一、MDR/IVDR实施后,NB的三大系统性顽疾

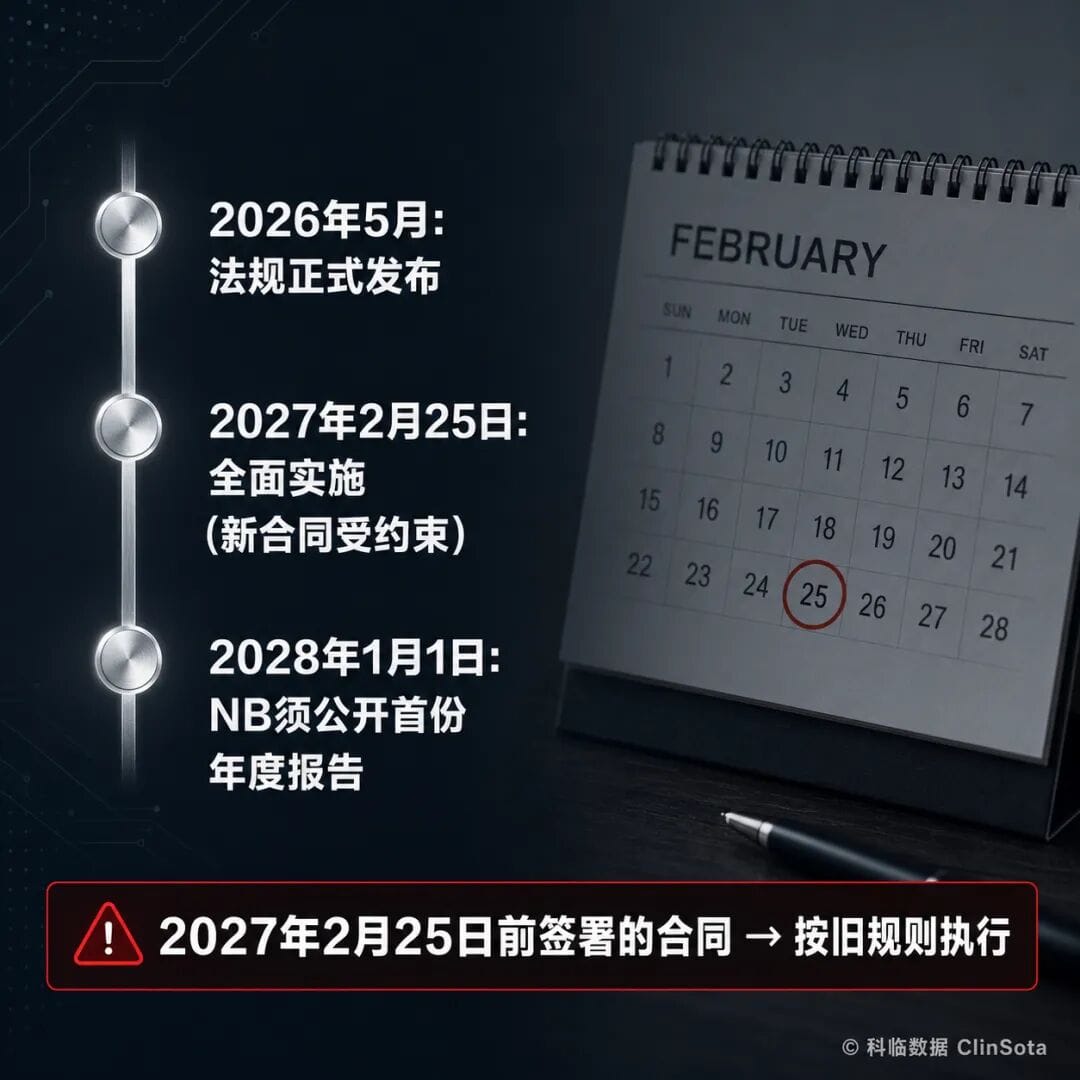
二、两个关键日期——千万别搞错
三、报价彻底透明化——告别“天价账单”和隐性收费
四、史上首次“硬性时间轴”——认证不再无限期拖延
申请审查与合同签署:30天(从收到完整申请起算) QMS审核:120天(从启动审核计划首个活动起算) 技术文档评估(产品验证):90天(从启动技术文档评估起算) 决策与发证:20天(从最后一次QMS或产品审查完成次日起算)

五、中断次数封顶——“Clock-stop”不能再无限拖
申请审查:最多1次 QMS审核:最多4次(多场地每增加1个,追加2次) 产品验证:最多4次 决策发证:最多1次

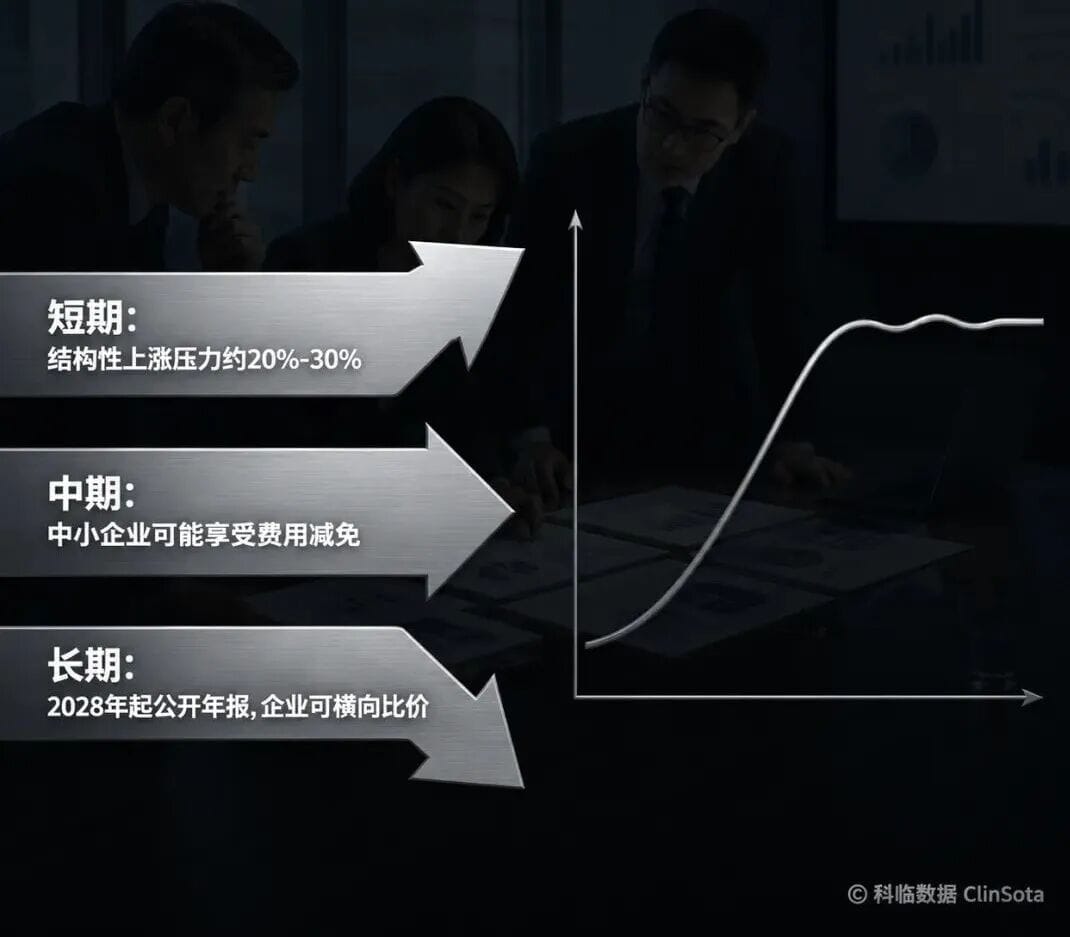
六、NB认证费用走向——短期上涨,中长期趋于合理
七、不同认证阶段,不同应对策略

八、医疗器械制造商的4项紧急准备

