


从CMEF看CGM全球注册策略与合规AI布局

一、全球CGM市场:增量时代的格局重塑
1. 市场规模与增长驱动力
糖尿病患病人数增加:世界卫生组织估计,全球糖尿病人数已接近8.3亿,低收入和中等收入国家占多数。这一庞大人群为CGM提供了长期需求基础。 技术迭代与消费化趋势:PPT强调CGM正在由专业医疗向大众健康转型,非处方(OTC)产品和订阅模式兴起,降低了用户入门门槛。无创/微创技术、AI预测算法等迭代提升了使用体验与精度。 医保和政策驱动:全球越来越多的医疗保险将CGM纳入报销范围,驱动市场渗透率提升。 以下是目前已获得NMPA注册证书的中国企业概览以供参考 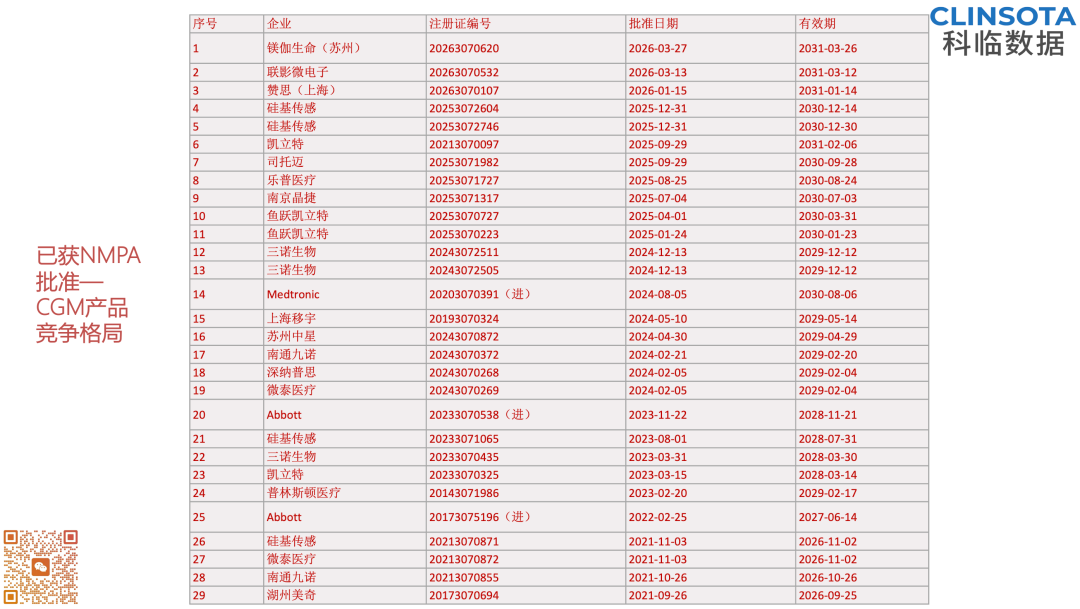
2. 竞争格局与变化
3. 技术趋势与生态化升级
产品消费化:从有创到微创再到无创技术,如近红外光谱或微针技术,逐步提升用户舒适度并拓展至更广泛的健康人群。 场景多元化:应用场景从家庭扩展到医院、社区和远程管理,并与胰岛素泵、AI算法等深度集成,形成闭环系统。 生态智能化:AI赋能使产品从数据监测工具升级为智能健康管理平台,提供预测性警报和个性化建议。 商业模式转变:订阅制、数据驱动服务和生态整合将成为主要盈利模式。
二、中欧法规差异:理解逻辑和路径的本质差异
1. 分类与注册单元
2. 注册路径与临床要求
3. 临床评价重点差异
三、中欧双报策略:一套流程、数据,识别GAP - 两国通行
注册单元对齐
全球多中心临床试验
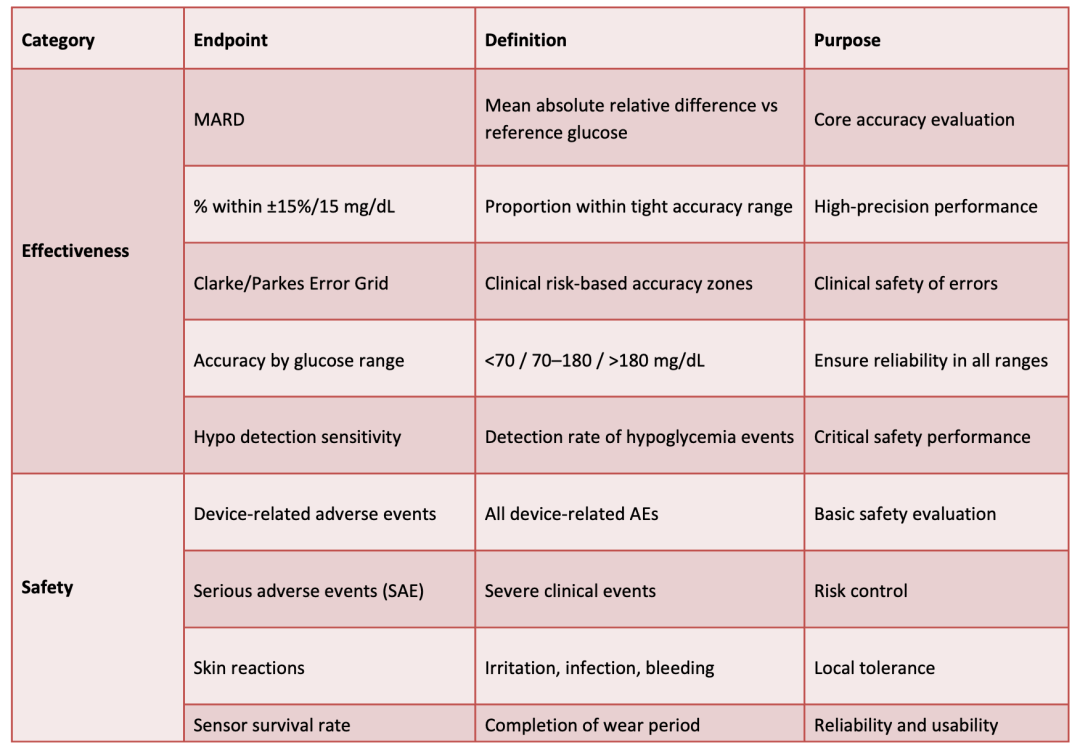
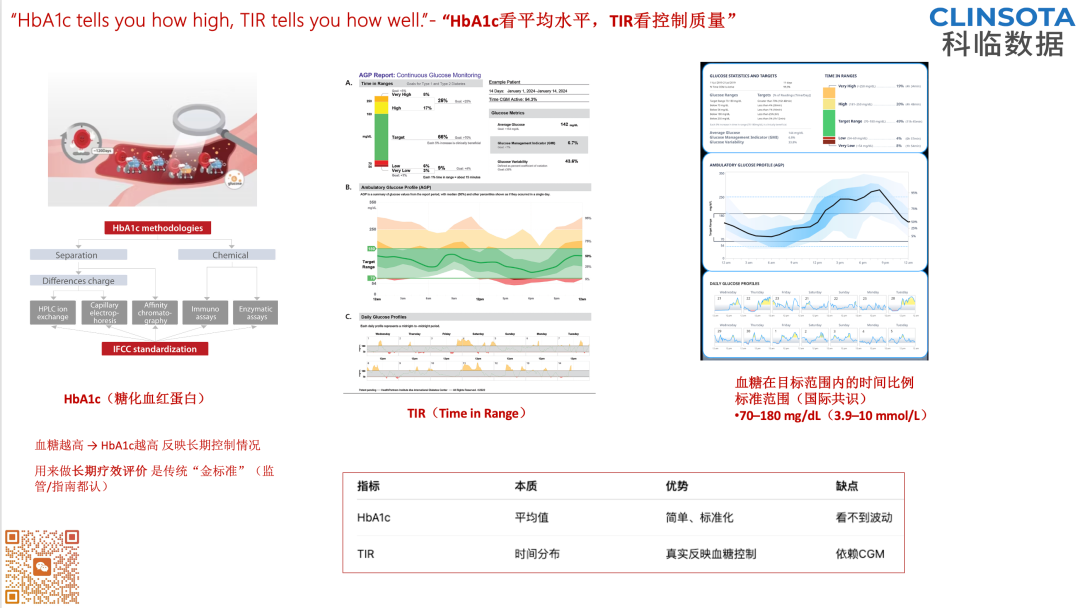
共用体系与文件
四、临床合规挑战:证据工程的复杂性
文献检索与SOTA构建繁琐:欧盟强调必须建立全面的SOTA,梳理治疗领域现状、替代方案、指标等。人工进行文献检索、筛选和分类耗时巨大,容易遗漏关键证据。 等同性评估难度大:MDR要求从技术特征、材料生物学特征和临床特征三方面证明申报器械与对比器械等同。缺乏充分论证会导致要求额外临床试验。 报告间一致性管理复杂:临床评价报告(CER)、临床评价计划(CEP)、PMCF计划和GSPR矩阵等文件相互关联。手工操作时,任何修改都可能导致其他文档不一致,增加返工风险。 监管环境变化快:欧盟MDR要求的风险管理、PMCF和安全更新(PSUR)是持续性的;中国也不断更新技术要求和指南。企业需要随时更新资料并保持符合性。 人才和成本压力:高质量合规团队稀缺,合规周期长、成本高,尤其对初创企业构成挑战。
五、AI合规平台:多智能体的落地实践
1. 多智能体协同
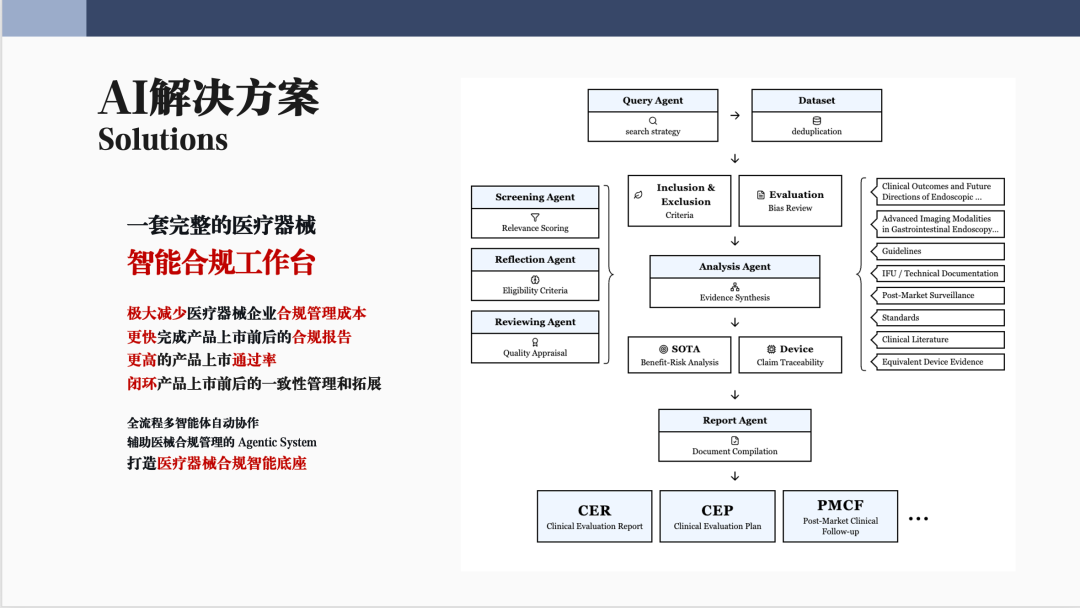
2. 闭环证据链与全产出对齐
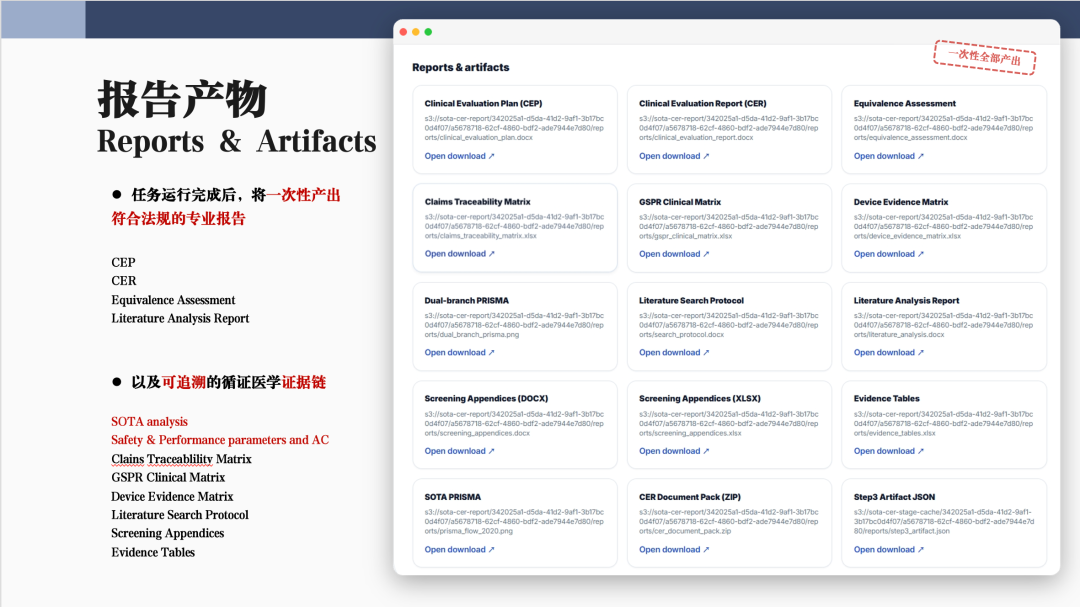
3. 等同性评估自动化
4. 降本提速与落地建议
时间节省:自动检索和报告生成显著减少人力投入,加快合规报告的完成速度。 降低成本:减少合规团队规模,缩短注册周期;避免由于文档不一致导致的返工。 提高质量:完整的证据链和自动对齐机制降低被Notified Body或NMPA补件的风险。 赋能中小企业:初创企业人手有限,通过平台可以快速搭建合规能力,专注于技术创新。
六、未来趋势与落地建议
1. 技术演进与市场拓展
2. 监管趋势与国际合作
3. 建议:合规即设计,数据即竞争力
结语


