


倒计时 6 个月!EUDAMED 强制使用在即,医疗器械企业必读
🔎什么是 EUDAMED
EUDAMED
Regulation (EU) 2017/745(MDR)及类似法规为欧盟医疗器械和 IVDR(体外诊断器械)构建了更加严格和透明的监管体系。对应地,European Database on Medical Devices(EUDAMED)被设计为整个系统的“数字中枢”——整合制造商/授权代表/进口商身份、器械注册 (UDI)/设备信息、公告机构与证书、市场监管、上市后警戒、临床研究等多个环节,实现从企业、产品到监管的全过程可追溯。
EUDAMED 包含 6 个模块:
“Actor / 参与者注册 (Actor Registration)”
“器械/UDI 注册 (UDI/Device Registration)”
“公告机构与证书 (Notified Bodies & Certificates)”
“市场监管 (Market Surveillance)”
“上市后监督 & 警戒 (Post-market surveillance & Vigilance)”
“临床调查 / 性能研究 (Clinical Investigation & Performance Studies)”
过去几年,这些模块逐步开放,只允许“自愿使用”;但随着监管推进,必须要合规。
最关键时间点
EUDAMED
根据欧盟官方 2025 年 11 月 27 日发布的通告(European Commission),EUDAMED 的头四个模块已被确认功能符合规范,并在官方公报 (OJEU) 发布。

这意味着,自 2026 年 5 月 28 日 起,以下四个模块将 强制使用:
Actor 注册
UDI/Devices 注册
Notified Bodies & Certificates
Market Surveillance
对于医疗器械企业 (制造商 / 授权代表 /进口商 /系统包生产商等) 而言,这不再是“可以选”的 — 而是进入或继续在欧盟市场销售产品的 硬性合规门槛。
因此,对于希望出口到欧洲,或在欧洲合法销售医疗器械 / IVD 产品的公司而言,现在到 2026-05-28 的这段 “过渡期 (transition period)” 是最后准备机会。
出口欧盟医疗器械企业的意义
EUDAMED
合规门槛提升:如果没有按时完成 Actor 注册 + UDI/器械注册 + NB/证书注册 + 市场监管信息备案,可能直接影响 CE 认证输出、产品出口、上市与销售。
全生命周期监管:EUDAMED 打通从“谁制造 / 谁授权 / 谁进口 + 产品是谁 + 认证机构 + 注册证书 + 上市后市场监测 / 警戒 / 回报 ”的全链路,透明度和可追溯性相比此前大幅提升。
行政合规风险增高:不按规定通过 EUDAMED 报备,可能导致市场准入障碍、监管处罚、声誉损失等。
时间窗口有限:距离 2026-05-28 只有 ~6 个月,对于尚未开始准备的企业,需要立即启动合规动作。
对中国医疗器械企业来说,这意味着:如果打算进入或继续通过欧盟合规 / CE 认证 / 欧盟市场准入,就必须将 EUDAMED 注册 / 数据梳理 / UDI 系统对接 / 合作公告机构 / 产品资料准备纳入近期重点工作。
应对措施 - 五大任务
EUDAMED
适用:制造商(含中国制造商)、欧代、进口商、系统包生产商。
企业要做什么?(10–20 分钟即可提交)
Step 1:准备材料
营业执照
制造商/欧代信息
PRRC 信息(合规负责人)
企业地址、联系人
Step 2:提交 Actor 注册
登录 EUDAMED → Actor 模块 → 上传资料。
Step 3:主管当局审核(3–10 天)
非欧盟企业由 欧代所在成员国 审核。
Step 4:获得 SRN
这是后续所有模块的“身份证
② 完成 UDI / 器械注册(最耗时的工作)
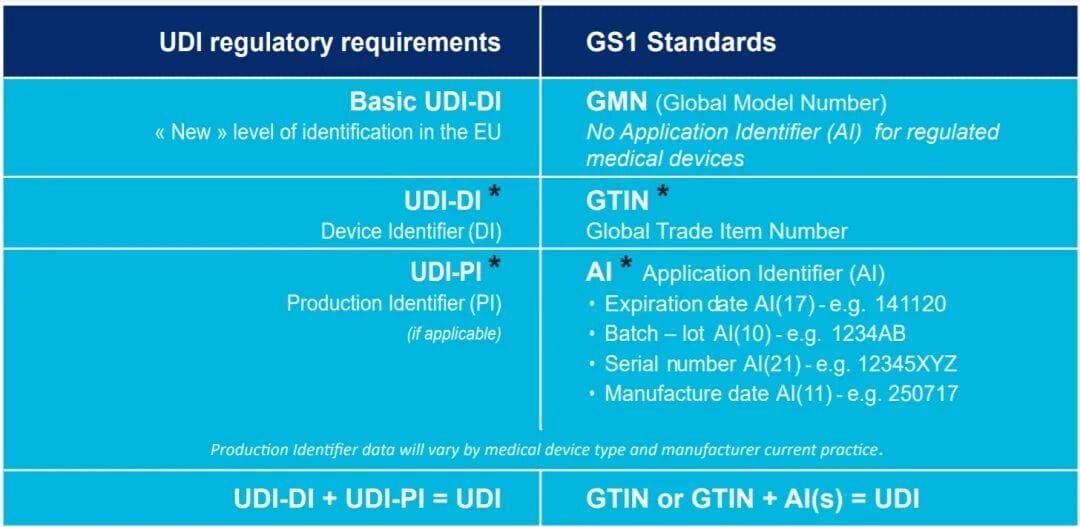
这是企业最容易低估的环节
Step 1:整理产品线列表(必做)
用一张 Excel 建立以下字段:
型号
风险等级
EMDN 编码
Basic UDI-DI
UDI-DI
包装层级
Step 2:申请/生成 UDI
通过 GS1 / HIBCC。
Step 3:在 EUDAMED Device 模块录入
录入以下必填字段:
Basic UDI-DI
UDI-DI
EMDN
产品用途
包装信息
制造商信息
Step 4:提交与保存记录
③ NB 证书登记(必须公告机构上传)
企业必须做的事
Step 1:确认证书覆盖的 Basic UDI-DI
否则 NB 无法上传。
Step 2:通知 NB 按时上传证书
企业 不能 自己上传,这是很多中国企业的误区。
Step 3:企业在系统内确认
确认 NB 上传的证书字段正确。
Step 4:定期检查证书有效期
续证、变更都必须同步。
④ 建立 EUDAMED 内部更新流程(强制后最关键)
企业必须建立 4 类流程:
1. 产品变更同步流程
型号、包装、标签、生产地址变化 → 必须更新。
2. 证书状态更新流程
续证、变更、扩证必须及时同步。
3. PMS / Vigilance(上市后)流程
不良事件、召回(FSCA)均需履行 MDR Article 87–100 要求。
4. 数据季度复核流程
确保 UDI、证书、产品数据一致。
⑤ 指定 PRRC + 制定 EUDAMED 项目时间表
这是很多企业忽略的部分
PRRC(合规负责人)职责(MDR Article 15)
审核所有 EUDAMED 数据
确保产品符合 MDR 要求
监督 PMS / Vigilance 数据
建议项目时间表(适用于 2025–2026)
2025-12:完成 SRN(Actor 模块) + 完成产品线总表2026-01:生成 Basic UDI-DI / UDI-DI + 准备 EUDAMED 字段2026-02:完成 ≥50% UDI / Device 录入2026-03:完成全部 NB 证书上传 + 企业确认2026-04:内部数据复核(UDI、Device、Certificates 全量核对)2026-05:PRRC 最终审核 → 全模块检查 → 5/28 强制上线
越早准备,越不容易踩点失败。
2026 年 5 月 28 日之后,所有欧盟出口企业必须遵守。

