

医疗器械临床试验的“全球GCP标准” ISO14155: 2026 正式发布
保护人体受试者、使用者或其他人员的权利、安全和福祉;
确保临床研究的科学性以及临床研究结果的可信度;
明确申办者和主要研究者的责任;
为参与医疗器械符合性评估的申办者、研究者、伦理委员会、监管机构及其他机构提供指导。
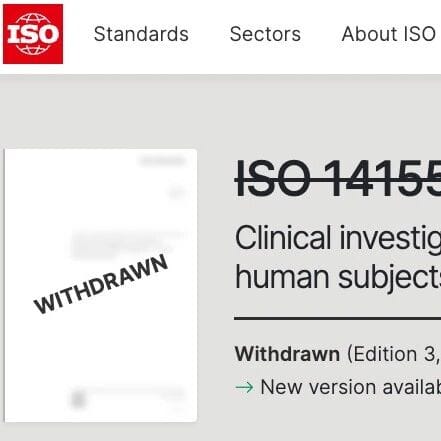
Table of Contents | Title |
Foreword | |
Introduction | |
1 | Scope |
2 | Normative references |
3 | Terms and definitions |
4 | Summary of good clinical practice principles |
5 | Ethical considerations |
5.1 | General |
5.2 | Improper influence or inducement |
5.3 | Compensation and additional health care |
5.4 | Registration in publicly accessible database |
5.5 | Responsibilities |
5.6 | Communication with the ethics committee |
5.7 | Vulnerable populations |
5.8 | Informed consent |
6 | Clinical investigation planning |
6.1 | General |
6.2 | Risk management |
6.3 | Justification for the design of the clinical investigation |
6.4 | Clinical investigation plan |
6.5 | Investigator's brochure |
6.6 | Case report forms |
6.7 | Monitoring plan |
6.8 | Investigation site selection |
6.9 | Agreement(s) |
6.10 | Labelling |
6.11 | Data monitoring committee |
6.12 | Clinical events committee |
7 | Clinical investigation conduct |
7.1 | General |
7.2 | Investigation site initiation |
7.3 | Investigation site monitoring |
7.4 | Adverse events and device deficiencies |
7.5 | Clinical investigation documents and documentation |
7.6 | Additional members of the investigation site team |
7.7 | Subject privacy and confidentiality of data |
7.8 | Document and data control |
7.9 | Investigational device accountability |
7.10 | Accounting for subjects |
7.11 | Auditing |
8 | Suspension, termination and close-out of the clinical investigation |
8.1 | Completion of the clinical investigation |
8.2 | Suspension or premature termination of the clinical investigation |
8.3 | Routine close-out |
8.4 | Clinical investigation report |
8.5 | Risk assessment and conclusions |
8.6 | Document retention |
9 | Responsibilities of the sponsor |
9.1 | Clinical quality management |
9.2 | Clinical investigation planning and conduct |
9.3 | Outsourcing of duties and functions |
9.4 | Communication with regulatory |
10 | Responsibilities of the principal investigator |
10.1 | General |
10.2 | Qualification of the principal investigator |
10.3 | Qualification of investigation site |
10.4 | Communication with the EC |
10.5 | Informed consent process |
10.6 | Compliance with the CIP |
10.7 | Medical care of subjects |
10.8 | Safety reporting |
Annex A | Clinical investigation plan |
A.1 | General |
A.2 | Identification and description of the investigational device |
A.3 | Justification for the design of the clinical investigation |
A.4 | Benefits and risks |
A.5 | Objectives and hypotheses |
A.6 | Design of the clinical investigation |
A.7 | Statistical design and analysis |
A.8 | Data management |
A.9 | Amendments to the CIP |
A.10 | Deviations from clinical investigation plan |
A.11 | Device accountability |
A.12 | Statements of compliance |
A.13 | Informed consent process |
A.14 | Adverse events |
A.15 | Vulnerable population |
A.16 | Suspension or termination |
A.17 | Publication policy |
A.18 | Bibliography |
Annex B | Investigator's brochure |
B.1 | General |
B.2 | Device information |
B.3 | Preclinical testing |
B.4 | Existing clinical data |
B.5 | Risk management |
B.6 | Regulatory references |
Annex C | Case report forms |
C.1 | General |
C.2 | Content and format |
C.3 | Procedural issues |
Annex D | Clinical investigation report |
D.1 | General |
D.2 | Cover page |
D.3 | Table of contents |
D.4 | Summary |
D.5 | Introduction |
D.6 | Methods |
D.7 | Results |
D.8 | Discussion |
D.9 | Terms |
D.10 | Ethics |
D.11 | Investigators |
D.12 | Signature |
D.13 | Annexes |
Annex E | Essential documents |
Annex F | Adverse event categorization |
Annex G | EC responsibilities |
Annex H | ISO 14971 application |
Annex I | Clinical development stages |
Annex J | Audits |
Annex K | Design considerations |
Bibliography |
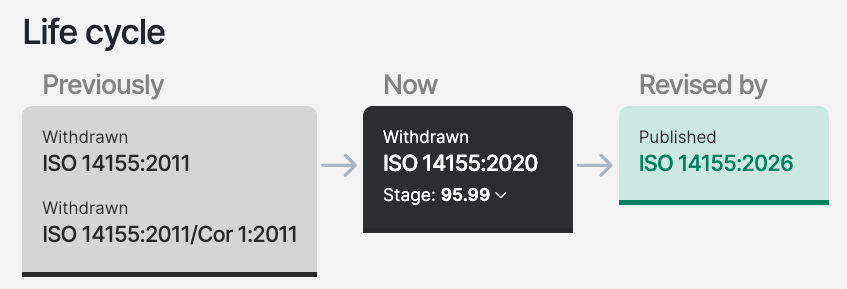
二、ISO 14155 第四版修订要点解读:临床试验治理正在从“合规完成”走向“风险精细化管理”
三、本次修订变化一览(基于原文变更清单)
序号 | 修订要点 |
1 | 修改了“临床性能(3.12)”的定义。 |
2 | 澄清了不允许偏离受试者资格标准;如需变更,应通过临床试验计划(CIP)修订处理(见 5.6.4)。 |
3 | 澄清了在适用情况下,应由受试者的法定指定代表取得知情同意(见 5.8.1)。 |
4 | 澄清了应给予受试者与他人(如家庭成员)讨论是否参与研究的机会(见 5.8.2)。 |
5 | 通过明确区分两类风险来澄清风险管理:一类是与器械使用有关的风险,另一类是由 CIP 要求、但并非常规临床实践组成部分的程序所带来的风险(见 6.2.1)。 |
6 | 新增了对残余风险进行评估的要求(见 6.2.2)。 |
7 | 更正了与试验器械使用相关风险的引用表述(见 6.2.1、7.4.4、8.2、附录 F、附录 H 及 3.2)。 |
8 | 将原先位于附录 A 的部分要求移入 6.4。 |
9 | 新增要求:数据监测委员会(DMC)应确认暂停或停止临床试验的条件(见 6.11)。 |
10 | 新增“临床事件委员会(CEC)”章节(见 3.8、6.12 及 A.14)。 |
11 | 澄清了可适用不良事件报告要求降低的情形(见 7.4.2)。 |
12 | 新增对由 CIP 要求之临床程序相关风险的管理要求(见 7.4.5)。 |
13 | 结合更新后的 7.4.4 和图 H.1,进一步澄清临床试验暂停或提前终止的流程(见 8.2)。 |
14 | 更新了 CIP 中“程序”章节,补充变量评估、记录和分析的方法与时点,并新增设备校准要求(见 A.6.4)。 |
15 | 澄清了非劣效界值和缺失数据处理的要求(见 A.7)。 |
16 | 新增要求:若不设 DMC,应说明其合理性(见 A.14)。 |
17 | 新增要求:受试者随访和持续照护应包括那些不同于常规实践的内容(见 A.16)。 |
18 | 澄清了“本地代表”相关内容,以更好地与国家监管要求协调一致(见 9.2.1)。 |
19 | 新增植入卡要求(见 9.2.2)。 |
20 | 将关于研究目标和研究设计的一般性要求归入 6.4(见 A.5)。 |
21 | 更新了不良事件分类,并在图 F.1 中澄清术语。 |
22 | 根据 6.2.1 更新了附录 H,并同步更新图 H.1。 |
23 | 引入了 estimand(估计目标)及其属性的原则(见 6.4、A.5、A.6、A.7 及附录 K)。 |
24 | 新增了注意事项(见 B.5)、试验器械使用培训信息(见 B.2)以及 in-silico 测试(见 B.3)。 |
25 | 新增“与器械缺陷相关的不良事件”这一类别——图 F.1 与图 F.2 现均适用。 |
四、以下是对主要更新或修改的核心内容总结:
4.1 版本前言中的“主要变化”
修改了“临床性能(3.12)”的定义。 澄清了不允许偏离受试者资格标准;如需变更,应通过临床试验计划(CIP)修订处理(见 5.6.4)。 澄清了在适用情况下,应由受试者的法定指定代表取得知情同意(见 5.8.1)。 澄清了应给予受试者与他人(如家庭成员)讨论是否参与研究的机会(见 5.8.2)。 通过明确区分两类风险来澄清风险管理:一类是与器械使用有关的风险,另一类是由 CIP 要求、但并非常规临床实践组成部分的程序所带来的风险(见 6.2.1)。 新增了对残余风险进行评估的要求(见 6.2.2)。 更正了与试验器械使用相关风险的引用表述(见 6.2.1、7.4.4、8.2、附录 F、附录 H 及 3.2)。 将原先位于附录 A 的部分要求移入 6.4。 新增要求:数据监测委员会(DMC)应确认暂停或停止临床试验的条件(见 6.11)。 新增“临床事件委员会(CEC)”章节(见 3.8、6.12 及 A.14)。 澄清了可适用不良事件报告要求降低的情形(见 7.4.2)。 新增对由 CIP 要求之临床程序相关风险的管理要求(见 7.4.5)。 结合更新后的 7.4.4 和图 H.1,进一步澄清临床试验暂停或提前终止的流程(见 8.2)。 更新了 CIP 中“程序”章节,补充变量评估、记录和分析的方法与时点,并新增设备校准要求(见 A.6.4)。 澄清了非劣效界值和缺失数据处理的要求(见 A.7)。 新增要求:若不设 DMC,应说明其合理性(见 A.14)。 新增要求:受试者随访和持续照护应包括那些不同于常规实践的内容(见 A.16)。 澄清了“本地代表”相关内容,以更好地与国家监管要求协调一致(见 9.2.1)。 新增植入卡要求(见 9.2.2)。 将关于研究目标和研究设计的一般性要求归入 6.4(见 A.5)。 更新了不良事件分类,并在图 F.1 中澄清术语。 根据 6.2.1 更新了附录 H,并同步更新图 H.1。 引入了 estimand(估计目标)及其属性的原则(见 6.4、A.5、A.6、A.7 及附录 K)。 新增了注意事项(见 B.5)、试验器械使用培训信息(见 B.2)以及 in-silico 测试(见 B.3)。 新增“与器械缺陷相关的不良事件”这一类别——图 F.1 与图 F.2 现均适用。
4.2 导言(Introduction)
4.3 范围(Scope)
保护受试者、使用者或其他人员的权利、安全和福祉; 确保临床试验的科学实施以及临床试验结果的可信性; 界定申办方和主要研究者的职责; 协助申办方、研究者、伦理委员会、监管机构以及参与医疗器械符合性评定的其他机构开展工作。
