


关于可植入器械和免除临床试验的 III 类器械清单的规定

1.授权法案的背景
授权法案的背景是关于成熟技术(WET)是相对简单的设备,具有常见和 设计稳定、变化不大,具有众所周知的安全性和临床性能特征以及在市场上的悠久历史。识别WET的标准已在医学领域制定设备协调小组指南(MDCG 2020-6)。 《(EU) 2017/745号条例》第61(6)(b)条列出了一系列技术,这些技术凭借对于WET,可能会受益于简化方法,在这种情况下,可免除进行临床研究的要求。此外,第61(8)条赋予了委员会将WET清单扩展到与所列设备类似的设备。 根据到目前为止在MDR方面积累的经验,本授权法案旨在扩大 《欧盟条例(EU)2017/745》第61(6)(b)条中规定的设备类型清单,以使其免于开展临床研究的要求。
2.法案通过前的程序
本授权法案所包含的WET设备清单是于2025年通过广泛咨询相关利益相关方制定的,这些利益相关方包括拥有MDCG临床调查与评估性能研究及评估工作组、 borderline与分类工作组观察员身份的人员,以及MDCG自身的主管当局成员。
3.授权法案的法律要素
该法律行为旨在扩大《欧盟法规(EU)2017/745》第 61(6)(b)条中可植入设备和 III 类设备的类型清单,这些设备豁免于进行临床试验的要求。

4. 判定为成熟器械WET 的核心考量:
具有通用、简单和稳定的设计
具有众所周知的安全性和临床性能已验证
具有众所周知的临床性能特点,它们是护理标准设备适应症和技术水平的演变;
在联合市场有较长的使用历史
SOTA一致性
5. 本次法规扩张WET器械范围
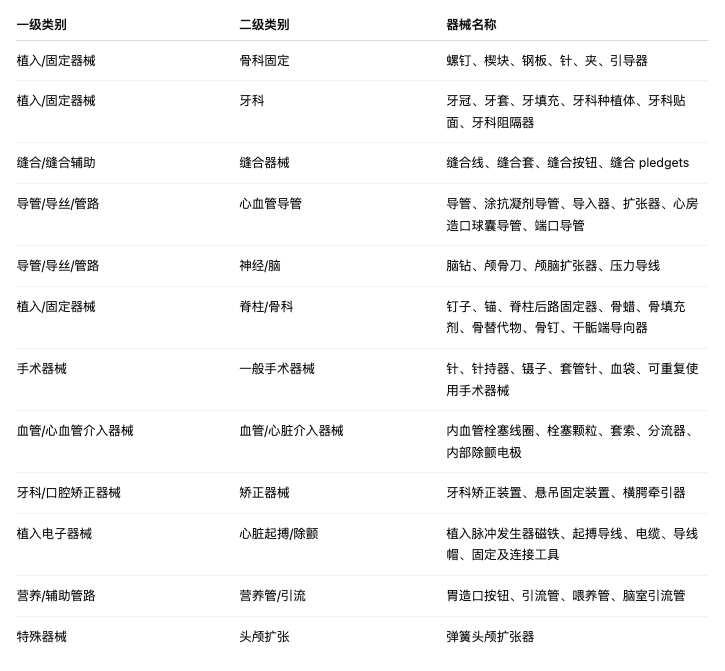
本条例应在《欧盟官方公报》上公布后的第二十日生效。本条例具有完全约束力,并直接适用于所有成员国。
6. 法规监管趋势分析
7. 企业临床合规策略布局考量
循证医学证据、 PMCF数据、 Registry 、RWE(真实世界数据)、临床评价(CER)地位被进一步强化,数据分析要更强、更系统、更标准化,从有没有临床试验数据” → “数据是否可信、完整、可追溯”。
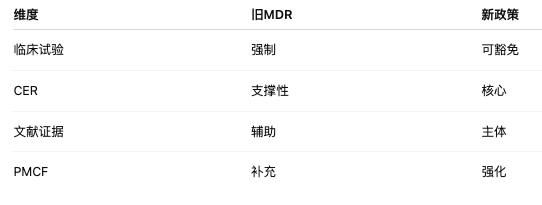
8. 结论或影响
欧盟监管体系有望正在从“临床试验驱动”全面转向“证据驱动”,企业核心能力不再是“做试验”,而是:构建高质量临床证据体系的能力。
